单细胞分析工具--ECAUGT提取hECA数据

基于先前的hECA文献笔记:hECA—人类单细胞表达图谱平台,学习使用python工具ECAUGHT高效提取特定类型的人类单细胞图谱数据。值得注意的是hECA对不同来源数据集仅进行了测序文库的标准化以及log转换,用户可根据特定应用场景进行适当的批次校正处理。
- 官方教程:http://eca.xglab.tech/ecaugt/index.html
pip install ECAUGT
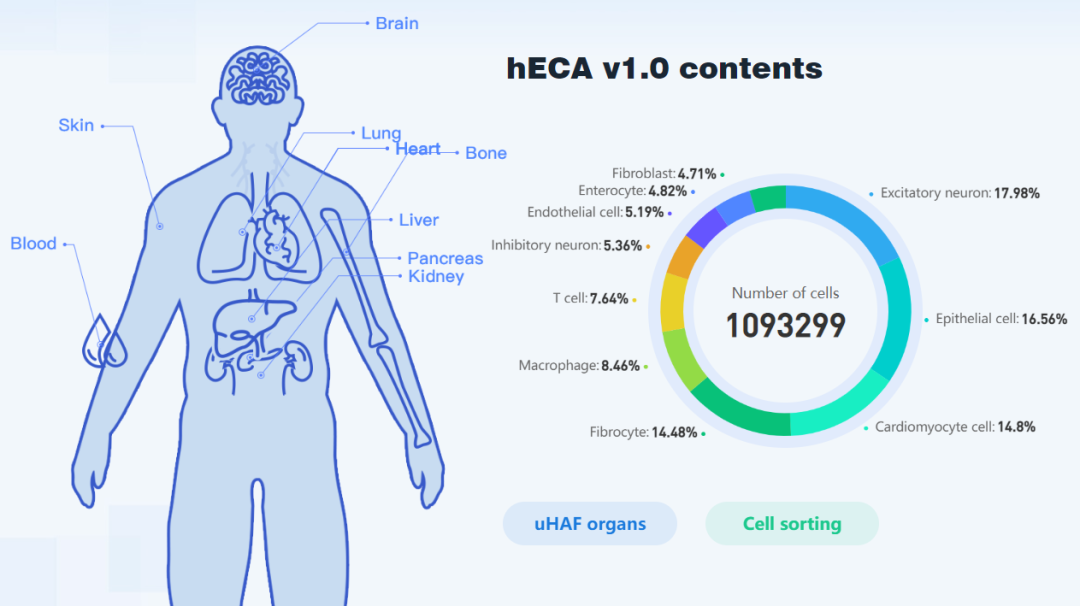
1、前期准备
## 加载包
import sys
import pandas as pd
import ECAUGT
import time
import multiprocessing
import numpy as np
## 建立API链接
# set parameters
endpoint = "https://HCAd-Datasets.cn-beijing.ots.aliyuncs.com"
access_id = "LTAI5t7t216W9amUD1crMVos" #enter your id and keys
access_key = "ZJPlUbpLCij5qUPjbsU8GnQHm97IxJ"
instance_name = "HCAd-Datasets"
table_name = 'HCA_d'
# setup client
ECAUGT.Setup_Client(endpoint, access_id, access_key, instance_name, table_name)
2、表型筛选
hECA数据储存方式是行名是细胞id,列名包括基因名(43878) + 表型信息(18),共43896列的巨大矩阵。
首先可根据表型信息(meta.data)筛选目标细胞群,常用的两个条件是器官(organ)与细胞(cell_type)类型
- 所支持的器官类型 http://eca.xglab.tech/#/organGallery
- 所支持的细胞类型 http://eca.xglab.tech/#/cellTypeList?viewType=AZ
## 如下查询肺组织的T细胞
rows_to_get = ECAUGT.search_metadata("organ == Lung && cell_type == T cell")
# 14894 cells found
rows_to_get[:5]
# [[('cid', 2000932)],
# [('cid', 2000962)],
# [('cid', 2000971)],
# [('cid', 2000978)],
# [('cid', 2000987)]]
## cell id格式为长度为2的元组,第一个元素是'cid',第二个元素是数字序号
## 常用逻辑操作符
# '==' means equal
# '<>' means unequal
# '&&' means AND operation
# '||' means OR operation
# '!' means not NOT operation
3、下载数据
df_result = ECAUGT.get_columnsbycell_para(
rows_to_get = rows_to_get[:20], ## 返回指定细胞id
cols_to_get=None, ## 返回指定基因/表型列
col_filter=None, ## 特定基因表达模式
do_transfer = True, ## 是否返回DataFrame格式
thread_num = multiprocessing.cpu_count()-1)
df_result.shape
# (20, 43896) 43878个基因信息 + 18列表型信息(metadata)
genes = df_result.columns[:43878] # 基因名
metaCols = df_result.columns[43878:43878+18] # 表型名
# Index(['cell_id', 'cell_type', 'cl_name', 'donor_age', 'donor_gender',
# 'donor_id', 'hcad_name', 'marker_gene', 'organ', 'original_name',
# 'region', 'sample_status', 'seq_tech', 'study_id', 'subregion',
# 'tissue_type', 'uHAF_name', 'user_id'],
# dtype='object')
meta = df_result.loc[:,metaCols]
meta.reset_index(inplace=True)
expr = df_result.loc[:,genes]
expr.reset_index(inplace=True)
expr=expr.drop(['cid'], axis=1)
- 筛选特定基因表达模式,并返回指定列的细胞数据
gene_condition = ECAUGT.set_gene_condition("PTPRC > 0.1 && CD3D>=0.1")
df_result = ECAUGT.get_columnsbycell_para(
rows_to_get = rows_to_get[:20],
cols_to_get=['CD3D','PTPRC','donor_id','hcad_name'],
col_filter=gene_condition,
do_transfer = True,
thread_num = multiprocessing.cpu_count()-1)
df_result.shape
# (5, 4)
4、分批下载
- 若目标细胞群过多,可分成小批量多次下载
from tqdm import tqdm
import pickle
for chunk in tqdm(range(int(1+len(rows_to_get)/500)),ncols=80):
# split batches
lb, rb = chunk*500, (chunk+1)*500
rows = rows_to_get[lb:rb]
if len(rows)<=0:break
# download rows from the unified Giant Table (uGT)
result = ECAUGT.get_columnsbycell_para(rows_to_get = rows,
cols_to_get = None, # download all columns
col_filter = None,
do_transfer = True,
thread_num = 24)
result.to_pickle("__temp_%d_%d.pk"%(lb,rb))
#print("downloading %d~%d"%(lb, rb))
#print(len(rows))
# load split batches
giant_table_list = []
for chunk in tqdm(range(int(1+len(rows_to_get)/500)),ncols=80):
lb, rb = chunk*500, (chunk+1)*500
fname = "__temp_%d_%d.pk" % (lb, rb)
with open(fname,'rb') as f:
df=pickle.load(f)
giant_table_list.append(df)
giant_table= giant_table_list[0]
for i in range(1, len(giant_table_list)):
giant_table = pd.concat([ giant_table, giant_table_list[i] ])
# remove intermediate results
del giant_table_list
import gc
gc.collect()
#giant_table.to_pickle("sorted_tcells_raw.pk")
df_result = giant_table
df_result.shape
# (14894, 43896)本文参与?腾讯云自媒体分享计划,分享自微信公众号。
原始发表:2023-02-02,如有侵权请联系?cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录