RNAseq|oncoPredict 药物反应预测,+基因,+分型,+模型 的联合可视化

RNAseq|oncoPredict 药物反应预测,+基因,+分型,+模型 的联合可视化

oncoPredict 是一款用来预测药物反应的R包,背景知识有很多介绍的了,这里介绍下真实的使用场景 以及 后续联合基因表达,分子分型 或者 预后模型等的联合。
一 载入数据,R包
1,安装R包
oncoPredict是CRAN中的包,直接install.packages安装,但是大概率会遇到缺少数据库相关R包的情况,根据提示安装即可。
install.packages("oncoPredict")
#BiocManager::install("GenomicFeatures")
#BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene")
library(oncoPredict)
2,输入数据
oncoPredict需要3个输入数据:(1)训练集表达数据-GDSC2 expression data (2)训练集药物反应数据-GDSC2 pType data (3)待预测的表达数据-our expression data 。
oncoPredict包提供了两种当前经典的药物预测训练集-Cancer Therapeutics Response Portal (CTRP-V2) 和 Genomics of Drug Sensitivity in Cancer (GDSC-V2) 。可以直接使用R包oncoPredict整理好的这两个数据库的rdata文件,下载链接https://osf.io/c6tfx/files/osfstorage。

CTRP可能是测序方式,提供了 FPKM和TPM数据。GDSC应该是芯片,提供了 RMA Normalized and Log Transformed。
3,读取三个数据集
训练集使用GDSC-V2 ,预测集使用之前使用的SKCM的表达矩阵
## 药物训练集
trainingExprData=readRDS(file='./DataFiles/Training Data/GDSC2_Expr (RMA Normalized and Log Transformed).rds')
dim(trainingExprData) #
trainingExprData[1:4,1:4]
#IMPORTANT note: here I do e^IC50 since the IC50s are actual ln values/log transformed already, and the calcPhenotype function Paul #has will do a power transformation (I assumed it would be better to not have both transformations)
trainingPtype<-exp(trainingPtype)
trainingPtype[1:4,1:4]
## 转录组数据
load("RNAseq.SKCM.RData")
fpkm[1:4,1:4]
fpkm_in <- fpkm %>%
column_to_rownames("gene") %>%
as.matrix()
testExprData = fpkm_in
testExprData[1:4,1:4]
trainingPtype是所有细胞系的不同药物的反应矩阵,trainingExprData是对应细胞系基因的表达矩阵,二者的样本名称是对应的。
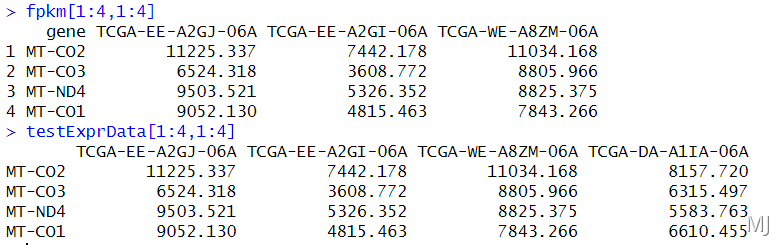
testExprData是待预测的自己的基因表达矩阵数据,行名为基因,列为样本。
二 预测药物反应
使用calcPhenoty函数进行预测。
1,配置参数
calcPhenotype的参数较多,没啥特殊需求建议使用默认的 ,参数解释相见官方脚步的参数说明:https://github.com/cran/oncoPredict/blob/master/R/CALCPHENOTYPE.R
batchCorrect<-"eb"
powerTransformPhenotype<-TRUE
removeLowVaryingGenes<-0.2
removeLowVaringGenesFrom<-"homogenizeData"
minNumSamples=10
selection<- 1
printOutput=TRUE
pcr=FALSE
report_pc=FALSEcc=FALSE
rsq=FALSE
percent=80
2,calcPhenotype计算
calcPhenotype的结果会默认输出在当前目录下的calcPhenotype_Output文件夹,结果输出为DrugPredictions.csv文件。
多次分析的话 注意覆盖原结果的问题。
calcPhenotype(trainingExprData=trainingExprData,
trainingPtype=trainingPtype,
testExprData=testExprData,
batchCorrect=batchCorrect,
powerTransformPhenotype=powerTransformPhenotype,
removeLowVaryingGenes=removeLowVaryingGenes,
minNumSamples=minNumSamples,
selection=selection,
printOutput=printOutput,
pcr=pcr,
removeLowVaringGenesFrom=removeLowVaringGenesFrom,
report_pc=report_pc,
cc=cc,
percent=percent,
rsq=rsq)
#读取结果
resultPtype <- read.csv('./calcPhenotype_Output/DrugPredictions.csv', header = T ,stringsAsFactors = F ,check.names = F)
resultPtype[1:4, 1:4]
# Camptothecin_1003 Vinblastine_1004 Cisplatin_1005
#1 TCGA-EE-A2GJ-06A 0.355799775 0.03555260 51.93174
#2 TCGA-EE-A2GI-06A 0.009369448 0.02313505 11.29654
#3 TCGA-WE-A8ZM-06A 0.083040399 0.04688820 76.53759
#4 TCGA-DA-A1IA-06A 0.152475833 0.38765839 17.42450如上就得到了每个样本的所有药物反应的结果。
三 基因+,分型+,预后+
既然得到的是每个样本的药物反应结果,当然就可以进行各种联合分析,这里分别以 目标基因 和 分子分型 进行示例。
1,重点基因表达量-相关性点图
重点基因可以来自于RNAseq|WGCNA-组学数据黏合剂,代码实战-一(尽)文(力)解决文献中常见的可视化图 找到的hub基因,RNAseq|Lasso构建预后模型,绘制风险评分的KM 和 ROC曲线构建的预后模型的基因,可以是单细胞scRNA分析|Marker gene 可视化 以及 细胞亚群注释--你是如何人工注释的?找到的celltype 差异基因,可以是RNAseq|免疫浸润也杀疯了,cibersoert?xCELL?ESTIMATE?你常用哪一个免疫浸润程度等等。
names(resultPtype)[1] <- "sample"
#以两个基因为示例
fpkm_gene_drug <- fpkm_in %>%
t() %>%
as.data.frame() %>%
dplyr::select(c("SPP1","TREM2")) %>%
rownames_to_column("sample") %>%
inner_join(resultPtype)
fpkm_gene_drug[1:4,1:4]
library(ggstatsplot)
ggscatterstats(
data = fpkm_gene_drug,
x = SPP1,
y = `BMS-754807_2171`,
xlab = "SPP1",
ylab = "BMS-754807_2171",
marginal = TRUE,
marginal.type = "densigram",
margins = "both",
xfill = "blue", # 分别设置颜色
yfill = "#009E73",
title = "Relationship between SPP1 and BMS-754807_2171",
messages = FALSE
) #BMS-754807_2171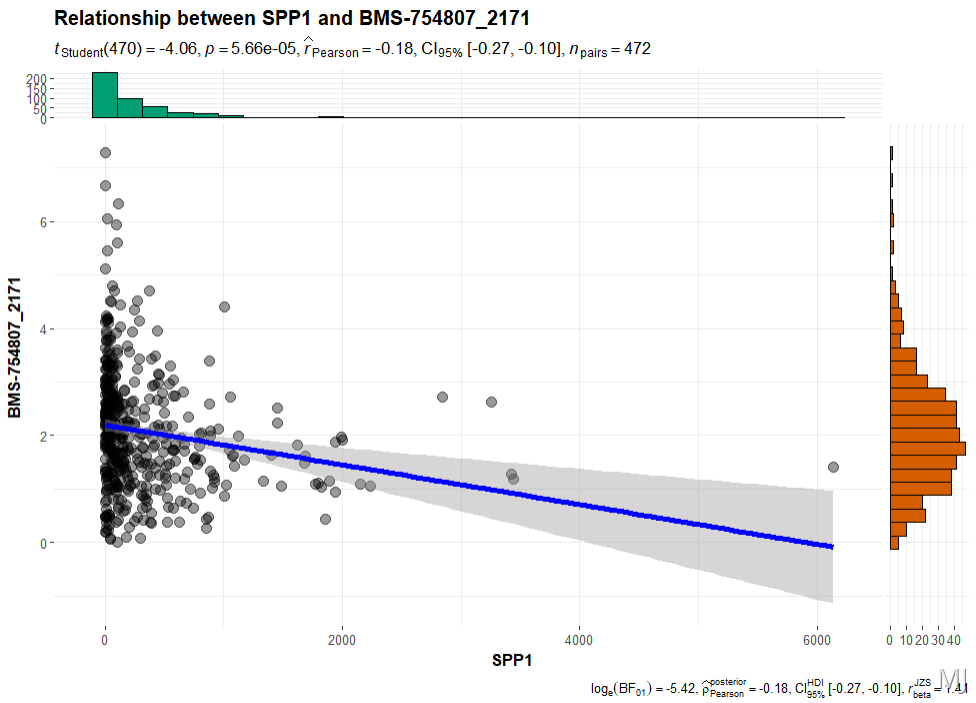
更多参数详见R|散点图+边际图(柱形图,小提琴图),颜值UP
2,聚类/预后模型-分组箱线图
分组性状可以来自于RNAseq|组学分型-ConsensusClusterPlus(一致性聚类), NMF(非负矩阵分解)的一致性聚类结果,可以是RNAseq|Lasso构建预后模型,绘制风险评分的KM 和 ROC曲线的风险分组,可以是关心的分类性状。
以无监督聚类结果为例
km <- read.csv("resultstrain.k=2.consensusClass.csv",header = T,stringsAsFactors = F ,check.names = F)
names(km) <- c("sample","cluster")
fpkm_km_drug <- km %>%
inner_join(resultPtype)
fpkm_km_drug[1:4,1:4]
ggboxplot(fpkm_km_drug, x="cluster", y="Cisplatin_1005", width = 0.6,
color = "black",#轮廓颜色
fill="cluster",#填充
palette = "npg",
xlab = F, #不显示x轴的标签
bxp.errorbar=T,#显示误差条
bxp.errorbar.width=0.5, #误差条大小
size=1, #箱型图边线的粗细
legend = "right") #图例放右边 +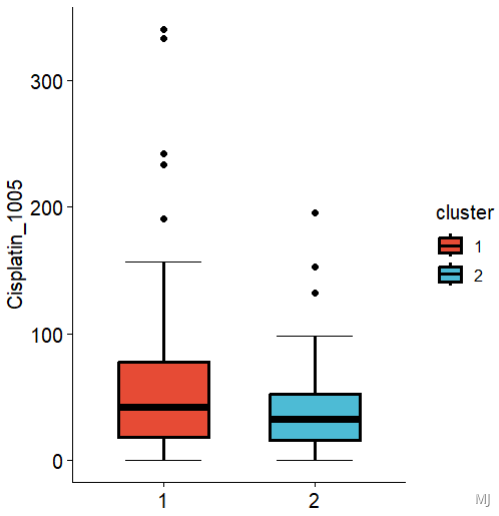
更多参数详见scRNA分析|自定义你的箱线图-统计检验,添加p值,分组比较p值
根据课题需要自行结合更多的分析方式。