scRNA|ComplexHeatmap自定义单细胞转录组celltype-level 热图可视化
scRNA|ComplexHeatmap自定义单细胞转录组celltype-level 热图可视化

一 载入R包,数据
使用之前注释过的sce.anno.RData数据 ,后台回复 anno 即可获取
library(ComplexHeatmap)
library(circlize)
library(tidyverse)
library(Seurat)
library(scater)
load("sce.anno.RData")
head(sce2,2)
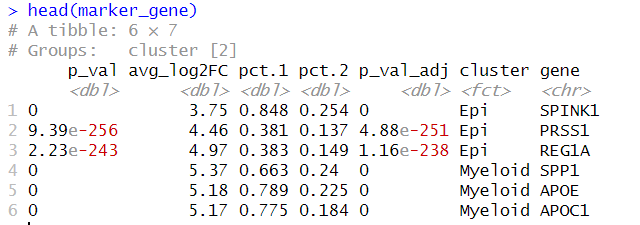
#计算marker 基因
all_markers <- FindAllMarkers(object = sce2)
marker_gene <- all_markers %>% group_by(cluster) %>% top_n(3, avg_log2FC)
head(marker_gene)
二 ComplexHeatmap 可视化
1,构建Heatmap 主体
首先将Seurat转为SingleCellExperiment ,然后利用aggregateAcrossCells 函数获取各celltype的平均表达矩阵 ,通过subset.row函数指定待计算均值的基因。可以通过
A:colorRamp2 自定义基因表达量的颜色;
B:这里选择经典marker基因展示,也可以是Findmarker找到的marker gene等;
sce <- as.SingleCellExperiment(sce2)
col_exprs <- colorRamp2(c(0,1,2,3,4),
c("#440154FF","#3B518BFF","#20938CFF",
"#6ACD5AFF","#FDE725FF"))
# cell type markers
marker_sign <- c("CD3E", 'CD3D', 'EPCAM', 'CD4', 'CD8A','SPP1', 'CD19', 'COL1A1', 'IGLC1')
celltype_mean <- aggregateAcrossCells(as(sce, "SingleCellExperiment"),
ids = sce$celltype,
statistics = "mean",
use.assay.type = "counts",
subset.row = marker_sign
)
celltype_mean@assays@data@listData$counts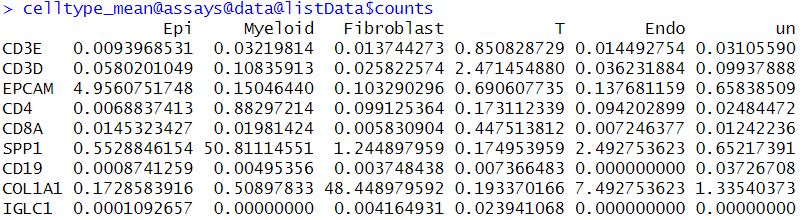
注:该功能类似于Seurat的AverageExpression 函数,在之前的多个推文中scRNA分析|单细胞GSVA + limma差异分析-celltype分组?样本分组?使用过了。
2,基础热图绘制
h_type <- Heatmap(t(assay(celltype_mean, "counts")),
column_title = "type_markers",
col = col_exprs ,
name= "mean exprS",
show_row_names = TRUE,
show_column_names = TRUE)
h_type
3,添加"天马行空"注释
注意SingleCellExperiment中的colData(celltype_mean) 存储的为类似seurat的metadata信息,这里熟悉下SingleCellExperiment的用法。
1) 添加celltype_mean中的细胞个数
anno <- colData(celltype_mean) %>%
as.data.frame %>%
select(celltype,ncells)
anno
# celltype ncells
#Epi Epi 9152
#Myeloid Myeloid 1615
#Fibroblast Fibroblast 2401
#T T 543
#Endo Endo 138
#un un 1612)meta信息中其他信息
首先group到celltype维度 ,然后计算每种celltype的 AUCell 均值。
AUCell <- colData(sce) %>%
as.data.frame() %>%
select(celltype, AUCell) %>%
group_by(celltype) %>%
summarise(AUCell = mean(AUCell)) %>%
as.data.frame() %>%
column_to_rownames("celltype")
AUCell
# AUCell
#Epi 0.1264182
#Myeloid 0.1067336
#Fibroblast 0.1288063
#T 0.1010705
#Endo 0.1283561
#un 0.10661683)计算每种celltype 有多少样本注释到
n_PID <- colData(sce) %>%
as.data.frame() %>%
select(celltype, sample) %>%
group_by(celltype) %>% table() %>%
as.data.frame() %>%
dplyr::filter(Freq>0) %>%
dplyr::group_by(celltype) %>%
dplyr::count(name = "sample") %>%
column_to_rownames("celltype")
n_PID
# sample
#Epi 16
#Myeloid 15
#Fibroblast 12
#T 15
#Endo 14
#un 104)计算不同celltype的sample/group 占比
indication <- unclass(prop.table(table(sce$celltype,sce$sample), margin = 1))
indication2 <- unclass(prop.table(table(sce$celltype,sce$group), margin = 1))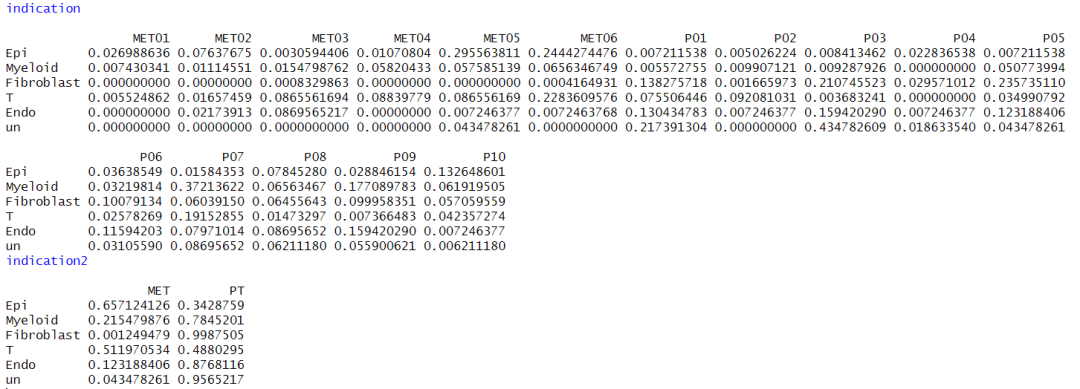
5)其他celltype的结果
这里以GSVA为例scRNA分析|单细胞GSVA + limma差异分析-celltype分组?样本分组?,其他可以转到celltype水平的结果均可以添加
library(msigdbr)
library(GSVA)
#选择基因集合
human_KEGG = msigdbr(species = "Homo sapiens", #物种
category = "C2",
subcategory = "KEGG") %>%
dplyr::select(gs_name,gene_symbol)#这里可以选择gene symbol或者ID
human_KEGG_Set = human_KEGG %>% split(x = .$gene_symbol, f = .$gs_name)#list
expr <- aggregateAcrossCells(as(sce, "SingleCellExperiment"),
ids = sce$celltype,
statistics = "mean",
use.assay.type = "counts"
)
gsva.kegg <- gsva(expr@assays@data@listData$counts,
gset.idx.list = human_KEGG_Set,
kcdf="Gaussian",
method = "gsva",
parallel.sz=1)
head(gsva.kegg)
4,构建注释信息
ComplexHeatmap可以自定义绝大多数的注释信息,信息非常多,这里简单介绍下 热图,柱形图,点图,色块几种常见的注释方式,更多的相见官网About | ComplexHeatmap Complete Reference (jokergoo.github.io)
1) GSVA结果热图
除了主体的热图外,还可以拼接其他celltype形式的图,这是使用GSVA热图示例。通过col设置颜色 ,使之颜色贴近主体的基因表达热图
h_state <- Heatmap(t(gsva.kegg[1:10,]),
column_title = "state_gsva",
#col = colorRamp2(c(-3,0,3), c("green","white","red"))
col = colorRampPalette(c("#440154FF", "#20938CFF", "#FDE725FF"))(10),
name= "gsva ",
show_row_names = TRUE,
show_column_names = TRUE)
h_state2)细胞类型注释
最常见的肯定是添加细胞类型的注释(分类变量),可以自定义颜色,尽量和umap图颜色一致
my36colors <-c('#53A85F', '#F1BB72', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175', '#E5D2DD', '#E0D4CA', '#F3B1A0'
)
col.list <- list(celltype = c(Epi = "#53A85F",
Myeloid = "#F1BB72",
Fibroblast = "#D6E7A3",
T = "#57C3F3",
Endo = '#476D87',
un = '#E95C59'
))
# Create HeatmapAnnotation objects
ha_anno <- HeatmapAnnotation(celltype = anno$celltype,
border = TRUE,
gap = unit(1,"mm"),
col = col.list,
which = "row")3) 其他信息注释
上述相关的统计变量均可以进行注释,通过width设置该部分注释图的宽度。
ha_meta <- HeatmapAnnotation(n_cells = anno_barplot(anno$ncells, width = unit(10, "mm")),
n_PID = anno_barplot(n_PID, width = unit(10, "mm")),
AUCell = anno_points(AUCell, width = unit(10, "mm")),
indication = anno_barplot(indication,width = unit(20, "mm"),
gp = gpar(fill = my36colors)),
indication2 = anno_barplot(indication2,width = unit(10, "mm"),
gp = gpar(fill = my36colors)),
border = TRUE,
annotation_name_rot = 90,
gap = unit(1,"mm"),
which = "row")5, 绘制 复杂 热图
将前面各部分的注释通过+号连接即可 ; 通过legend添加图例信息
# Create HeatmapList object
h_list <- h_type +
h_state +
ha_anno +
ha_meta
# Add customized legend for anno_barplot()
lgd <- Legend(title = "celltype_sample",
at = colnames(indication),
legend_gp = gpar(fill = my36colors))
lgd2 <- Legend(title = "celltype_group",
at = colnames(indication2),
legend_gp = gpar(fill = my36colors))
# Plot
draw(h_list,annotation_legend_list = list(lgd,lgd2))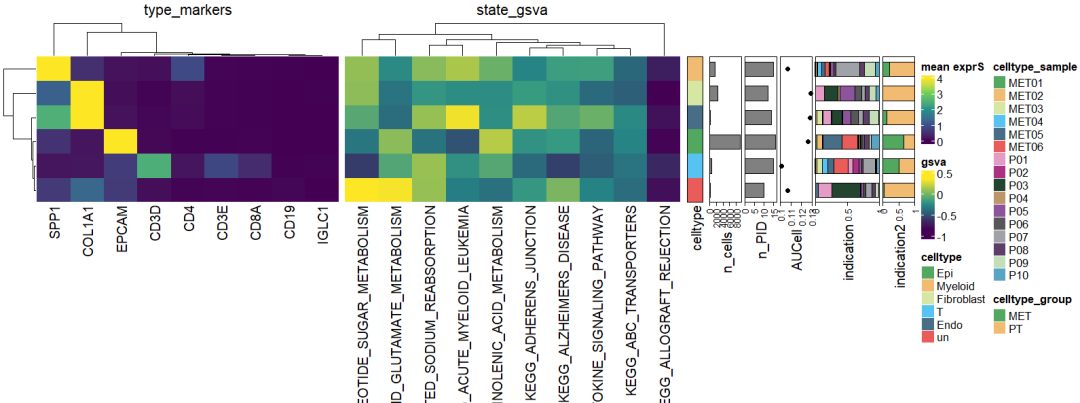
OK,最终得到如上所示的图,其他celltype level的注释均可以添加上去。
参考资料:
About | ComplexHeatmap Complete Reference (jokergoo.github.io)