全基因组 - 人类基因组变异分析(PacBio) (4)-- DeepVariant
原创全基因组 - 人类基因组变异分析(PacBio) (4)-- DeepVariant
原创
一、SNPs和INDELs变异检测
单核苷酸多态性(Single Nucleotide Polymorphism,SNP)指的是基因组中单个核苷酸腺嘌呤(A)、胸腺嘧啶(T)、胞嘧啶(C)或鸟嘌呤(G)在物种成员之间或个体配对染色体之间的差异, 是最常见也最简单的一类造成基因组多样性的DNA序列变异。
插入缺失(insertion-deletion,InDel),这里一般指小于50bp的变异,即在DNA序列中添加或删除少量碱基,主要指在基因组某个位置上发生较短长度的线性片段插入(Insert)或者缺失(Deletion)的现象,合称为InDel。
我们对下机数据进行比对分析 (pbmm2软件),提取全基因组中所有的潜在多态性SNP位点和小片段插入/缺失InDel位点(DeepVariant软件),后期再根据质量值、深度、重复性等因素做进一步的过滤筛选,最终得到高可信度的SNP和InDel数据集并注释(1)。
SNP和INDEL变异检测有助于我们更深入地了解基因组,生物性状的表现,物种的起源与进化,认识基因变异和疾病的之间的联系。从测序数据中进行准确的变异检测也是生物学、医学研究和精准医学的基础我们对下机数据进行比对分析 (pbmm2软件),提取全基因组中所有的潜在多态性SNP位点和小片段插入/缺失InDel位点(DeepVariant软件),后期再根据质量值、深度、重复性等因素做进一步的过滤筛选,最终得到高可信度的SNP和InDel数据集并注释(1)。
我们对下机数据进行比对分析,提取全基因组中所有的潜在多态性SNP位点和小片段插入/缺失InDel位点,再根据质量值、深度、重复性等因素做进一步的过滤筛选,最终得到高可信度的SNP数据集并注释。
二、变异检测工具
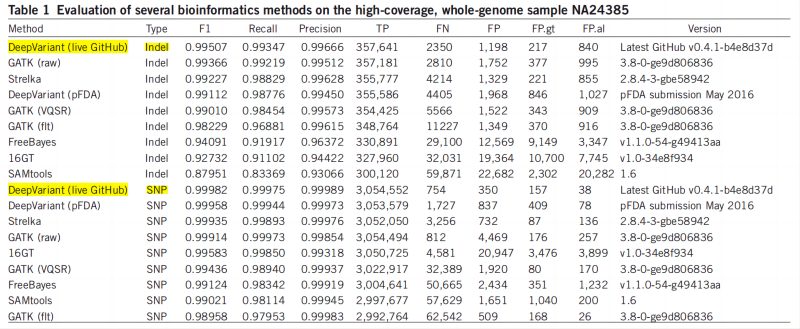
目前SNP和INDEL变异检测的软件有很多,比如老牌行业“金标” 由Broad Institute 开发 GATK,即Genome Analysis Toolkit 基因组分析工具。在变异软件综合评测中(2,3),DeepVariant软件在三代测序数据中表现是非常优秀的 (图1,图2,图3)。

PacBio生信分析培训推荐DeepVariant作为SNP和INDEL变异检测的软件,并且对于小型变异检测PacBio官方推荐的也是DeepVariant(图4), 所以接下来我们详细介绍下DeepVariant的使用方法。

三、DeepVariant简介
官方网址:https://github.com/google/deepvariant
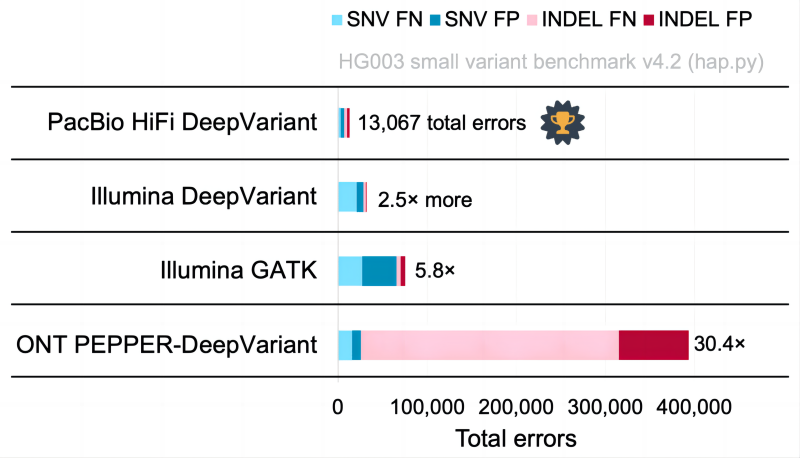
DeepVariant是由谷歌Google基于深度卷积神经网络开发的一款从DNA测序数据中快速较精确识别碱基变异位点的软件(4)。这个工具在准确率上和精确度上,比传统的比对拼接方法都高出一大截。DeepVariant,把工作量巨大的拼接问题(高通量测序碎片化的结果拼接成完整的基因序列),转变成了一个典型的图像分类问题。而图像分类正是谷歌擅长的技术。在2016 PrecisionFDA的Truth Challenge比赛中,DeepVariant获得了最高SNP性能奖,PacBio +DeepVariant(Highest SNP Performance)(图5)。在那之后,Google Brain团队又将错误率降低了50%。
DeepVarient软件运行运行流程如下图6所示:
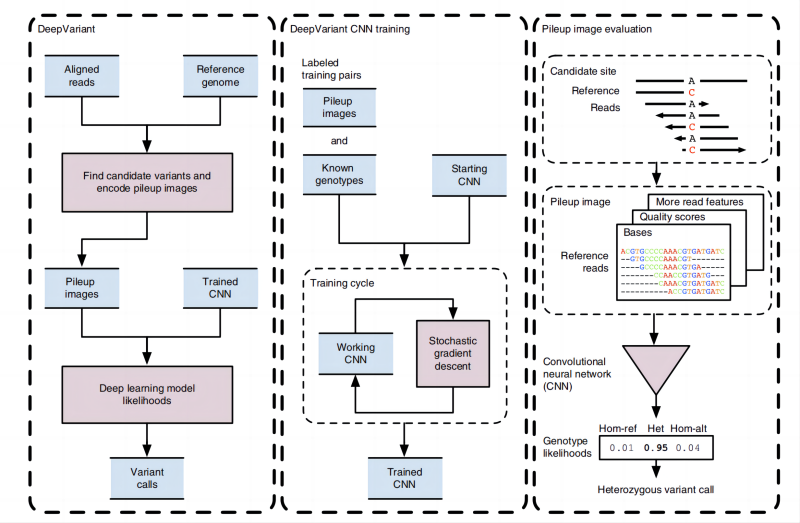
左边:筛选候选的变异位点集合;中间:SNN训练样本;右边:用训练好的模型判断Genotype
四、DeepVariant安装及使用
1. 软件安装
DeepVarient官方提供了3种安装方式:
- Docker,官方推荐使用的安装方式。
- 源代码构建。
- 二进制文件。
使用官方推荐安装方式pull docker镜像
#指定DeepVariant版本,这次安装为最新版本v1.6.0, 2023年10月24日发布。
$ BIN_VERSION="1.6.0"
#拉取docker镜像,大小为5.74GB
$ docker pull google/deepvariant:"${BIN_VERSION}"2. 数据准备
- 样本参考基因组文件
例如上一节pbmm2用到的GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz.
参考基因组需要samtools进行索引
#如果没有安装samtools,可以使用conda进行安装。
$ conda install -c bioconda samtools
#GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz 已经重命名为GRCh38.fa
#samtools进行索引
$ cd Human_ref
$ samtools faidx GRCh38.fa
#查看索引后的文件
$ ls
# GRCh38.fa GRCh38.fa.fai- 比对排序后的BAM文件
例如, 经过pbmm2比对,排序,索引后的bam文件:
HG002_1.align.bam HG002_1.align.bam.bai HG003.align.bam HG003.align.bam.bai HG004.align.bam HG004.align.bam.bai
3. 运行DeepVariant
#使用方法
$ BIN_VERSION="1.6.0"
$ docker run \
-v "YOUR_INPUT_DIR":"/input" \
-v "YOUR_OUTPUT_DIR:/output" \
google/deepvariant:"${BIN_VERSION}" \ #根据DeepVariant版本号来设置
/opt/deepvariant/bin/run_deepvariant \
--model_type=WGS \ #根据应用替换为其中的一种[WGS,WES,PACBIO,ONT_R104,HYBRID_PACBIO_ILLUMINA] WGS和WES为二代Illumina数据
--ref=/input/YOUR_REF \ #samtools索引的参考基因组
--reads=/input/YOUR_BAM \ #比对过的bam文件
--output_vcf=/output/YOUR_OUTPUT_VCF \ #输出的vcf
--output_gvcf=/output/YOUR_OUTPUT_GVCF \ #输出的gvcf, 用于合并样本进行变异分析
--num_shards=$(nproc) \ #CPU核数
--logging_dir=/output/logs \ #每一步的日志文档
--haploid_contigs="chrX,chrY" #如果用GRCh38则设置为"chrX,chrY",GRCh37则设置为"X,Y"。
# --regions "chr20:10,000,000-10,010,000" 可以指定变异检测范围HG002_1,HG003,HG004实际样本运行:
#HG002_1
$ nohup docker run \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/input" \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/output" \
google/deepvariant:"1.6.0" \
/opt/deepvariant/bin/run_deepvariant \
--model_type=PACBIO \
--ref=/input/Human_ref/GRCh38.fa \
--reads=/input/HG002_1.align.bam \
--output_vcf=/output/HG002_1.vcf.gz \
--output_gvcf=/output/HG002_1.gvcf.gz \
--num_shards=20 \
--logging_dir=/output/HG002_1_DeepV_logs \
--haploid_contigs="chrX,chrY" &
#HG003
$ nohup docker run \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/input" \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/output" \
google/deepvariant:"1.6.0" \
/opt/deepvariant/bin/run_deepvariant \
--model_type=PACBIO \
--ref=/input/Human_ref/GRCh38.fa \
--reads=/input/HG003.align.bam \
--output_vcf=/output/HG003.vcf.gz \
--output_gvcf=/output/HG003.gvcf.gz \
--num_shards=20 \
--logging_dir=/output/HG003_DeepV_logs \
--haploid_contigs="chrX,chrY" &
#HG004
$ nohup docker run \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/input" \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/output" \
google/deepvariant:"1.6.0" \
/opt/deepvariant/bin/run_deepvariant \
--model_type=PACBIO \
--ref=/input/Human_ref/GRCh38.fa \
--reads=/input/HG004.align.bam \
--output_vcf=/output/HG004.vcf.gz \
--output_gvcf=/output/HG004.gvcf.gz \
--num_shards=20 \
--logging_dir=/output/HG004_DeepV_logs \
--haploid_contigs="chrX,chrY" &
# nohup 挂起不间断
# &放入后台运行4.合并多个样本的gvcf文件
多样本联合变异(Joint Calling)需要用到 GLnexus工具。
GLnexus是由DNAnexus开发,用于可扩展的gVCF合并和联合变异(joint calling)要求群体测序项目,GL即genotype likelihood之意(5)。
GATK作为变异检测金标准软件,缺点在于速度很慢。尽管在分析上游的比对、单样本HaplotypeCaller检测等环节已经有很多替代品,如Sentieon、Parabricks等,但下游joint calling这一步优化仍然有限。而这正是整个GATK流程的最限速的步骤,在GATK中只能通过分区的方法来加速,效果非常有限(5)。
GLnexus的开发解决了这个痛点问题,在速度上不说几十上百倍的提升,至少也有十多倍。对于大规模群体/队列而言(主要针对人类基因组开发),是个非常好的工具(5)。
Deepvariant 和 Clara Parabricks 都推荐它来做联合变异(5)。
#下载GLnexus docker镜像
$ docker pull quay.io/mlin/glnexus:v1.2.7
#运行代码
$ sudo docker run \
-v "${DIR}":"/data" \
quay.io/mlin/glnexus:v1.2.7 \
/usr/local/bin/glnexus_cli \
--config DeepVariant \
--bed "/data/${CAPTURE_BED}" \
/data/HG004.g.vcf.gz /data/HG003.g.vcf.gz /data/HG002.g.vcf.gz \
| sudo docker run -i google/deepvariant:${VERSION} bcftools view - \
| sudo docker run -i google/deepvariant:${VERSION} bgzip -c \
> ${DIR}/deepvariant.cohort.vcf.gz帮助文档:
#查看帮助文档
$ docker run \
quay.io/mlin/glnexus:v1.2.7 \
/usr/local/bin/glnexus_cli --help
##帮助文档
Merge and joint-call input gVCF files, emitting multi-sample BCF on standard output.
Options:
--dir DIR, -d DIR scratch directory path (mustn't already exist; default: ./GLnexus.DB)
--config X, -c X configuration preset name or .yml filename (default: gatk)
--bed FILE, -b FILE three-column BED file with ranges to analyze (if neither --range nor --bed: use full length of all contigs)
--list, -l expect given files to contain lists of gVCF filenames, one per line
--more-PL, -P include PL from reference bands and other cases omitted by default
--squeeze, -S reduce pVCF size by suppressing detail in cells derived from reference bands
--trim-uncalled-alleles, -a remove alleles with no output GT calls in postprocessing
--mem-gbytes X, -m X memory budget, in gbytes (default: most of system memory)
--threads X, -t X thread budget (default: all hardware threads)
--help, -h print this help message
Configuration presets:
Name CRC32C Description
gatk 268790301 Joint-call GATK-style gVCFs
gatk_unfiltered 484625853 Merge GATK-style gVCFs with no QC filters or genotype revision
xAtlas 3445896276 Joint-call xAtlas gVCFs
xAtlas_unfiltered 920229760 Merge xAtlas gVCFs with no QC filters or genotype revision
weCall 2936345659 Joint-call weCall gVCFs
weCall_unfiltered 2838640462 Merge weCall gVCFs with no filtering or genotype revision
DeepVariant 2857227159 Joint call DeepVariant whole genome sequencing gVCFs
DeepVariantWGS 2857227159 Joint call DeepVariant whole genome sequencing gVCFs
DeepVariantWES 4105299981 Joint call DeepVariant whole exome sequencing gVCFs
DeepVariant_unfiltered 116393675 Merge DeepVariant gVCFs with no QC filters or genotype revision
Strelka2 3838963651 [EXPERIMENTAL] Merge Strelka2 gVCFs with no QC filters or genotype revisionHG002_1,HG003,HG004实际样本gvcf合并:
根据帮助文档 --config 可以设置为DeepVariant ,DeepVariant和DeepVariantWGS应该是一样的preset。如果使用GATK或其它软件进行变异分析则设置相应的模型
#joint-calling,最后得到deepvariant.cohort.vcf.gz
$ sudo docker run \
-v "/mnt/data/home/mli/Desktop/pb_WGS":"/data" \
quay.io/mlin/glnexus:v1.2.7 \
/usr/local/bin/glnexus_cli \
--config DeepVariant \
/data/HG002_1.gvcf.gz /data/HG003.gvcf.gz /data/HG004.gvcf.gz \
| docker run -i google/deepvariant:1.6.0 bcftools view - \
| docker run -i google/deepvariant:1.6.0 bgzip -c \
> deepvariant.cohort.vcf.gz
#未设置
--bed "/data/${CAPTURE_BED}" 五、结果说明
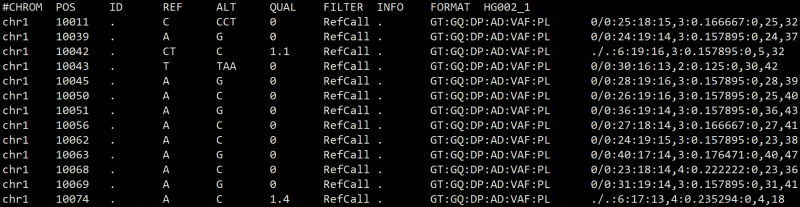
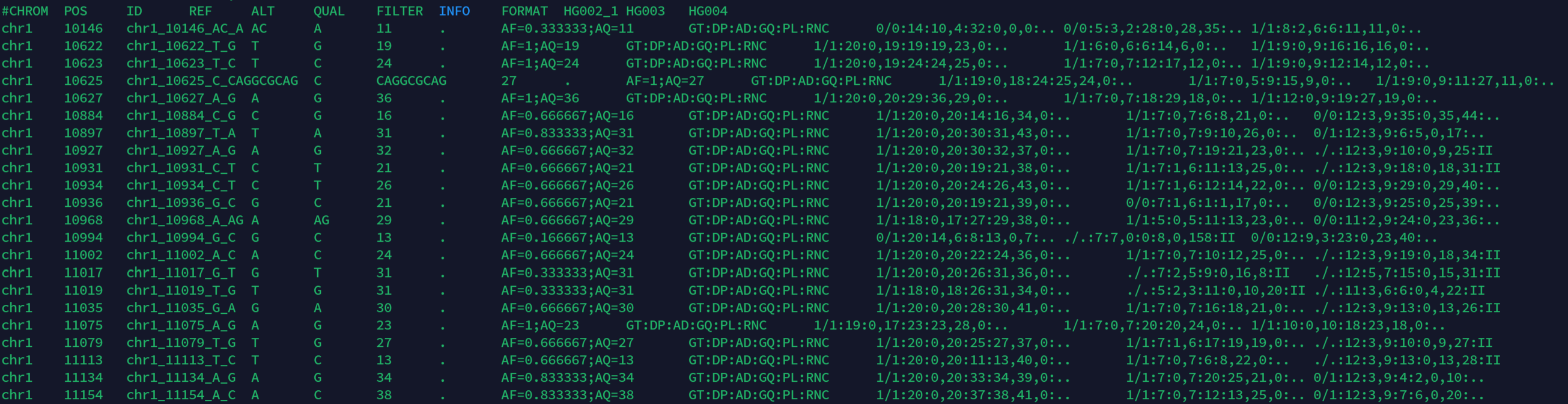
DeepVariant软件输出结果为vcf格式文件(图7),相信做生物信息的小伙伴都很熟悉了,这里不再赘述(1)。
HG002_1单个样本vcf:
HG002_1, HG003, HG004 三个样本Joint Calling的deepvariant.cohort.vcf.gz:
特别说明:
DeepVariant运行结束后,SNP和INDEL还在一个vcf文件里,如果为了后续单独分析,我们可以用GATK分离他们,命令如下(1):
$ gatk SelectVariants -R /input/YOUR_REF \
-V /input/YOUR_VCF \
-O /output/YOUR_RAW_SNP \
-select-type SNP|INDEL-select-type参数分别给定SNP和INDEL,将会分别得到对应变异类型的结果,输出仍然是vcf格式的文件。有了这个结果后,就可以进行后续的分析了。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。