J. Chem. Theory Comput. | AI驱动的柔性蛋白-小分子复合物建模
J. Chem. Theory Comput. | AI驱动的柔性蛋白-小分子复合物建模

今天为大家介绍的是来自陈语谦教授团队发表在Journal of Chemical Theory and Computation的论文,“Equivariant Flexible Modeling of the Protein?Ligand Binding Pose with Geometric Deep Learning”,博士生董铁君为第一作者。该文提出了一种新的AI驱动的蛋白-小分子复合物结构柔性建模方法FlexPose,可准确高效的预测复合物结构、亲和力及模型置信度。模型采用标量-矢量二元特征表示和 SE(3)-等变网络,以端到端动态建模复合物结构;同时提出构象感知的预训练和弱监督学习策略提高模型在未见过的化学空间中泛化性。在PDBbind,APObind数据集上的评估显示,在涉及蛋白质构象变化的任务中,模型的精度和效率大幅高于传统的分子对接及近期基于AI的方法。在基于数据集相似性的评价中,两个构象感知策略很大程度上提高了模型在低相似性化学空间中的性能。此外模型预测的亲和力及置信度估计为后续的药物研发提供了有效直接的参考。

模型架构
FlexPose 将一对蛋白质配体表示为由节点(蛋白质残基,小分子原子)和边组成的图(图 1b),其中每个节点和边都被初始化为标量(含其生化属性,如残基/原子类型)和一组欧几里得矢量集合(含其几何特征,如蛋白质原子间的相对位置)。与只包含标量特征的常用方法(只使用距离或扭转角来表示几何特征)不同,这种标量-矢量双重特征表示法允许模型明确管理节点的动态变化,而不是简单地依赖于其与所有相邻节点的相对位置。此外,双重特征使模型够覆盖更多的蛋白质残基,在无需直接访问其全原子结构的情况下拥有足够的几何表示。
如图1b 所示,FlexPose采用编码器-解码器式设计。蛋白质编码器和配体编码器首先将三维蛋白质结构和二维配体图分别嵌入潜在空间。然后,复合解码器通过多轮循环(图 1c)从头开始构建配体结构(从随机的高斯噪声开始),同时重建蛋白质结构。在每个循环中,复合解码器都会迭代更新特征和结构。简而言之,复合解码器对标量和矢量特征进行信息传递,其中每个节点通过汇总相邻节点的注意力加权信息来更新其特征,值得注意的是,更新结构和特征时,注意力权重是共享的。在每个循环结束时,复合解码器会对配体约束能量最小化,以确保结构的几何正确。然后如图1c 所示,特征和结构会到下一个循环进行进一步完善,额外的浅层网络用于估计亲和力。
为了提高模型的通用性,FlexPose用构象感知任务对编码器进行了预训练(图1d),同时该文提出了构象感知的弱监督学习(WSL,图1e),以扩展学习到的化学空间。构象感知预训练任务鼓励模型学习与化学性质相关(如屏蔽属性预测)的标量特征和与三维结构相关任务(如结构去噪)的矢量特征。这样的设计使解码器能够用更好的特征表示构建结构。构象感知WSL鼓励模型对未知结构的预测尽可能接近与其最相似的低置信度的结构。为减少潜在偏差,低置信度的结构最好由自下而上的方法生成,如分子力学等。当预测结构的化学空间与训练样本的化学空间几乎完全不重叠时,构象感知WSL将有助于模型建模新结构。
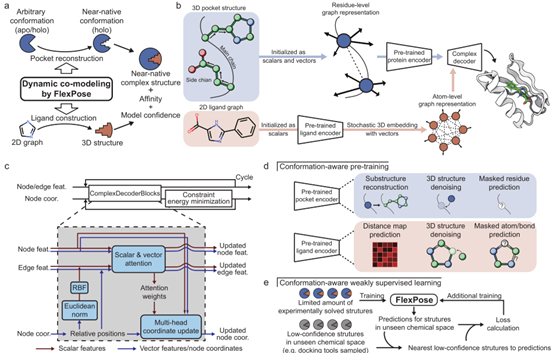
图 1
基于Apo蛋白预测复合物结构
文章首先评估了模型基于apo(native unbound)态蛋白预测原生复合物结构的能力,这对传统的对接方法来说极具挑战性。与apo构象相比,大多数holo(ligand-bound)构象显示出结构差异,通常是由于侧链构象的变化引起。传统的对接方法在结构变化时会面临严重的采样压力,此外许多对接工具只能将蛋白质视为刚性成分(半柔性对接)。文章使用了从 APObind和 PDBbind数据集中提取的两个测试集进行评估,apo-refined集(N = 937)和 apo-core集(N = 168)。
FlexPose的性能大大优于所有测试的传统对接工具(图 2c-f),在两个测试集中,预测构象与原生构象之间的均方根偏差(rmsd)值要小得多。图 2a,b 显示了两个样本,图 2c,e 显示了两个测试集的成功率。与表现最好的对接工具(RosettaLigand)相比,FlexPose在apo-refined和apo-core的成功率分别提高了 36.60% 和 40.48%。进一步将FlexPose 与最近的深度学习方法进行了比较,结果 FlexPose也取得了压倒性的优势,与排名第二的深度学习方法EDM-Dock相比,成功率提高了 33.41%。
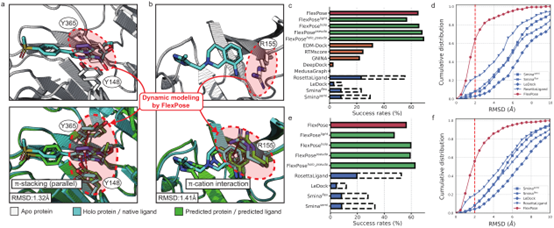
图 2
交叉对接与重对接任务评估
文章测试了 FlexPose 在交叉对接与重对接任务的能力。重对接是用原生复合物蛋白结构作为输入。交叉对接是用从不同配体得到的次优蛋白构象重现原生复合体结构。DBbind 数据集中的core set(N = 278)和cross-core set(N = 1305)上,将 FlexPose与14种传统方法或基于机器/深度学习的方法进行了比较。
如表 1 所示,在交叉对接任务中,FlexPose 的表现大大优于所有测试方法,在重对接任务中,FlexPose 的准确率也与最好的测试方法相当。在交叉对接方面,FlexPose及其变体的成功率在 0.644 到 0.735 之间,远高于排名第二的RTMscore方法(0.519)。结果表明,即使用holo态蛋白质,所有测试的对接工具和深度学习方法都显示出有限的能力,而 FlexPose实现了更好的准确性。
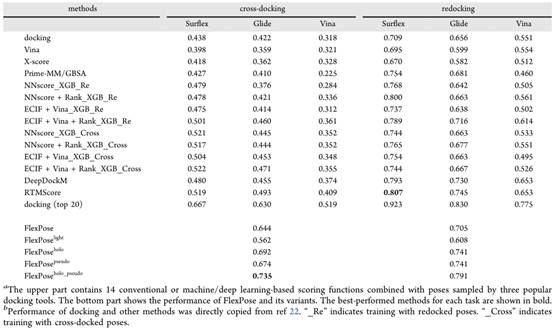
表 1
构象感知策略提高模型在unseen化学空间中预测能力
文章在交叉对接任务中测试了分子相似性的影响,训练集根据其与测试集的蛋白质和配体相似度进行筛选,同时比较了构象感知预训练和WSL在此类情况下的能力(图 3)。在构象感知预训练中,使用了额外的蛋白质和有机小分子来训练两个编码器(图 1d)。在预训练中没有使用天然复合物结构。相比之下,构象感知WSL通过使用未见化学空间中的低置信度复合物结构来对解码器进行训练(图1e)。WSL计算的是预测结构与其最近的低置信度结构之间的损失。
如图3所示,分子相似性对模型性能有影响,模型在使用预训练编码器或WSL后有明显改善,尤其是在相似性截断值较低的情况下。文章有两个主要发现:(1)蛋白质相似性比配体相似性的影响更大。当配体相似性临界值从1.0降到0.4时,基线模型(图 3b)的成功率从0.59降到0.39。然而当蛋白质相似性截止值降至0.4时,模型成功率降至0.20。这表明该模型善于推断已知蛋白质的新结合模式,但在预测未见过的蛋白质时却面临挑战,即使要预测的配体与训练集相似。(2)预训练的编码器和构象感知WSL都能在很大程度上提高模型性能(图 3c,d),而WSL的表现更好。如图3a-d所示,在大多数相似性截止条件下,两者都能提高成功率(最高可达0.14)。训练集越严格(即相似性截止值越低),提高的幅度就越大。例如,在蛋白质和配体的相似性截止值为1.0时,预训练编码器和WSL与基线相比分别提高了0.02和0.04;在蛋白质和配体的相似性截止值为0.4时,则分别可以提高0.06和0.14。
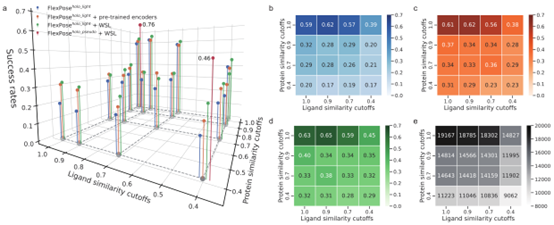
图 3
高效建模蛋白构象变化
FlexPose 可高效预测蛋白质的构象变化,重塑蛋白质侧链,并提供骨架CA和CB原子位置估计值。对于所有氨基酸类型,模型预测侧链构象的χ角误差(图 4a)都比 RosettaLigand低得多。如图2a,b所示,模型将参与相互作用的残基从apo构象重塑为holo构象。图 2a 中的红圈显示了apoY365与原生配体构象之间的冲突,预测结果改善了这一冲突,此外在预测中还建立了非共价相互作用(如π-π堆叠)。模型对骨架 CA 原子预测的平均误差分别为 0.78?和1.41?。上述结果表明,模型在重塑蛋白质构象变化方面具有显著的精确性。
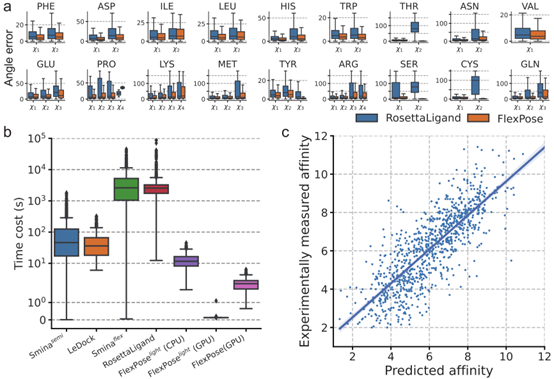
图 4
亲和力和模型置信度估计提供互作分析参考
FlexPose 提供了对结合亲和力和模型置信度的估计,这对蛋白质配体相互作用的后期分析非常有用。如图4c所示,模型对亲和力估计的皮尔逊相关系数(Pearson R)分别为0.809 和0.801。为了评估预测的可靠性,文章引入了一个称为 "ENS-factor "的模型置信度指标,它不需要额外的可训练参数。它基于这样一个假设:初始三维嵌入不同,模型都倾向于对有把握的部分做出一致的预测。ENS-factor的值是作为预测结构集合内偏差的简化表示来计算的。ENS-factor值越高,表明模型的可信度越高。图5显示了四个可视化样本。ENS-factor(即高置信度,以绿色显示)接近天然位置。相比之下,ENS-factor低(即置信度低,用红色表示)的子结构误差较大。模型在分子层级上的ENS-factor与rmsd(图 5e)之间的 Pearson R值为-0.61,原子层级的ENS-factor与原子位置误差(图 5f)之间的 Pearson R 值为-0.54。这些结果表明 ENS 因子可以代表预测中的潜在误差,为进一步的结构分析(如非共价相互作用)提供直接的视觉参考。

图 5
结论
FlexPose是一种高效的端到端深度学习框架,可用于柔性建模蛋白质配体复合物结构。得益于标量-向量双特征表示、SE(3)-等变网络、构象感知预训练和 WSL,其准确性大大超过了所有测试过的对接工具和近期深度学习方法,且大大缩短了预测时间。此外,模型能准确估计结合亲和力和模型置信度,以协助后期分析,这对于计算机辅助的药物相关开发(如药物设计、药物筛选)非常有用。
参考资料
Equivariant Flexible Modeling of the Protein–Ligand Binding Pose with Geometric Deep Learning; Tiejun Dong, Ziduo Yang, Jun Zhou, and Calvin Yu-Chian Chen; Journal of Chemical Theory and Computation Article ASAP
DOI: 10.1021/acs.jctc.3c00273
代码
https://github.com/tiejundong/FlexPose