scRNA|RΑφCytoTRACE v2¥”0ΩΣ ΦΆξ≥…ΒΞœΗΑϊΖ÷Μ·«±Ρή‘Λ≤β
scRNA|RΑφCytoTRACE v2¥”0ΩΣ ΦΆξ≥…ΒΞœΗΑϊΖ÷Μ·«±Ρή‘Λ≤β

CytoTRACE v2 ‘Ύ2024.03‘¬ΖΔ±μ‘Ύ‘Λ”Γ±ΨMapping single-cell developmental potential in health and disease with interpretable deep learningΓΘV2 Ι”ΟΩ…Ϋβ Ά–‘ΒΡAIΥψΖ®ά¥‘Λ≤βΒΞœΗΑϊRNA≤β–ρ ΐΨίΒΡœΗΑϊΖ÷Μ·«±ΡήΓΘ≥ΐΝΥΗχ≥ω¥”0Θ®Ζ÷Μ·Θ©ΒΫ1Θ®»ΪΡήΘ©ΒΡΝ§–χΖΔ”ΐ«±ΡήΕ»ΝΩΫαΙϊΆβΘ§ΜΙΗυΨίœΗΑϊΒΡΖΔ”ΐ«±ΡήΫχ––Ζ÷ΈΣ6άύΘΚΨΏ”–ΙψΖΚΖ÷Μ·«±ΡήΒΡ»ΪΡή(totipotent)ΚΆΕύΡή(pluripotent)Η…œΗΑϊΘ§ΒΫΡήΙΜ≤ζ…ζ≤ΜΆ§ ΐΝΩΒΡœ¬”ΈœΗΑϊάύ–ΆΒΡ ΤΉœΒœό÷Τ–‘ΕύΡήœΗΑϊΘ®lineage-restricted oligopotentΘ©Θ§ΕύΡή(multipotent)ΚΆΒΞΡή(unipotent)œΗΑϊΘ§‘ΌΒΫΉν÷’ΒΡ Ζ÷Μ·Θ®differentiatedΘ©œΗΑϊΓΘ
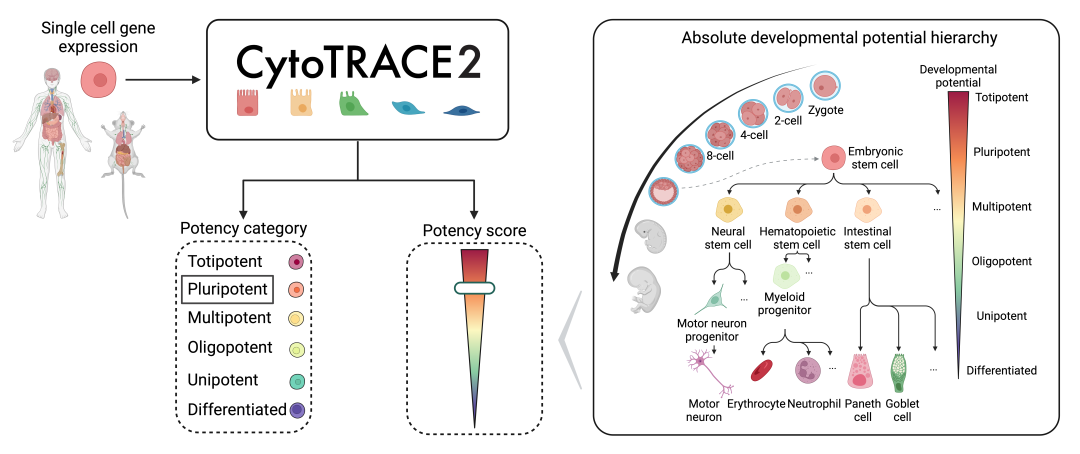
œύΫœV1ΒΡΙΠΡήΚΆάμ¬έΒΡΗΡΫχœξΦϊΈΡœΉ’ΐΈΡΘ§‘Ύ¥ζ¬κ Βœ÷…œCytoTRACE v2÷–≤πΖ÷ΈΣΝΥRΑφ±ΨΚΆPythonΑφ±ΨΘ§Α≤ΉΑRΑφ±ΨΒΡΜΑΈό–η≈δ÷ΟpythonΒΡΜΖΨ≥Θ§ Ι”ΟΟ≈Φς¥σΖυΫΒΒΆΓΘ
“Μ ‘Ί»κRΑϋΘ§ ΐΨί
1Θ§RΑϋΑ≤ΉΑ ΦΑ ΫβΨω±®¥μ
ΗυΨίhttps://github.com/digitalcytometry/cytotrace2?tab=readme-ov-file÷–ΒΡΖΫ ΫΫχ––Α≤ΉΑ
Θ®1Θ© Ι”Οdevtools::install_github÷±Ϋ”Α≤ΉΑ
devtools::install_github("digitalcytometry/cytotrace2", subdir = "cytotrace2_r")
library(CytoTRACE2)
# ≥ωœ÷±®¥μ
Using github PAT from envvar GITHUB_TOKEN
Downloading GitHub repo digitalcytometry/cytotrace2@HEAD
Error in utils::download.file(url, path, method = method, quiet = quiet, :
download from 'https://api.github.com/repos/digitalcytometry/cytotrace2/tarball/HEAD' failedΘ®2Θ©»γΙϊ≥ωœ÷…œ ωΒΡ±®¥μΘ§’β ±Κρ÷Μ“ΣΫΪ±®¥μΡΎ»ίΒΡΓΑhttps://api.github.com/repos/digitalcytometry/cytotrace2/tarball/HEADΓ± Η¥÷ΤΒΫΆχ÷ΖΥ―ΥςάΗΜΊ≥ΒΘ§ΨΆΜαœ¬‘Ί“ΜΗωΈΡΦΰtar.gzΒΡ―ΙΥθΈΡΦΰΘ§»ΜΚσΈ“Ο«‘Ό±ΨΒΊΑ≤ΉΑΦ¥Ω…ΓΘ
# ±ΨΒΊΑ≤ΉΑ
remotes::install_local("./digitalcytometry-cytotrace2-6fe2bad.tar.gz",
subdir = "cytotrace2_r", # ΧΊ βΒΡ
upgrade = F,dependencies = T)
library(CytoTRACE2)
library(tidyverse)
library(Seurat)ΉΔΘΚ¥ρΩΣtar.gz―ΙΥθΑϋΩ…“‘Ω¥ΒΫΉς’ΏΖ÷ΒΡpython ΚΆr Αφ±ΨΘ§Υυ“‘’βάο–η“Σ Ι”Οsubdir≤Έ ΐ÷ΗΕ®ΈΣcytotrace2_r ΓΘ
ΉΔΘΚΤδΥϊΒΡgithubΑϋ≥ωœ÷άύ–Ά±®¥μ“≤Ω…“‘ Ι”Ο…œ ωΖΫ ΫΫχ––ΫβΨωΘ§“ΜΑψ≤Μ–η“Σ…η÷Οsubdir ΓΘ
2Θ§ΉΦ±ΗΒΞœΗΑϊ ΐΨί
»ΜΚσ Ι”Ο÷°«ΑΉΔ ΆΙΐΒΡsce.anno.RData ΐΨί Θ§ΈΣΫΎ ΓΉ ‘¥Θ§ΟΩ÷÷œΗΑϊάύ–ΆΥφΜζ≥ι»Γ30%ΒΡ ΐΨίΓΘ
load("sce.anno.RData")
sce2@meta.data$CB <- rownames(sce2@meta.data)
sample_CB <- sce2@meta.data %>%
group_by(celltype) %>%
sample_frac(0.3)
sce3 <- subset(sce2,CB %in% sample_CB$CB)
sce3
# An object of class Seurat
Εΰ CytoTRACE v2 Ζ÷Έω
1Θ§CytoTRACE v2 Ζ÷Έω
ΗΟΑφ±ΨΩ…“‘Ϋ” ήΒΞœΗΑϊΕ‘œσ Μρ’Ώ ΒΞœΗΑϊΨΊ’σΒΡΝΫ÷÷–Έ ΫΘ§Έο÷÷Ω…“‘ «»ΥΜρ’Ώ–Γ σΘ®Ρ§»œΘ©ΓΘ±ΨΆΤΈΡ « Ι”Ο »Υ ΒΡΒΞœΗΑϊΕ‘œσΘ®sce3Θ©Ϋχ––cytotrace2Ζ÷ΈωΒΡ ΨάΐΓΘ
####### δ»κseurat Ε‘œσ###########
cytotrace2_result_sce <- cytotrace2(sce3,
is_seurat = TRUE,
slot_type = "counts",
species = 'human',
seed = 1234)
cytotrace2_result_sce
An object of class Seurat
51911 features across 4202 samples within 1 assay
Active assay: RNA (51911 features, 2000 variable features)
4 dimensional reductions calculated: pca, umap, tsne, harmonyδ»κΒΡ «ΒΞœΗΑϊΕ‘œσΘ§ΒΟΒΫΒΡ“≤ «ΒΞœΗΑϊΕ‘œσΘ§«“meta–≈œΔ÷–ΑϋΚ§ΝΥœύΙΊscoreΒΡΫαΙϊΓΘ
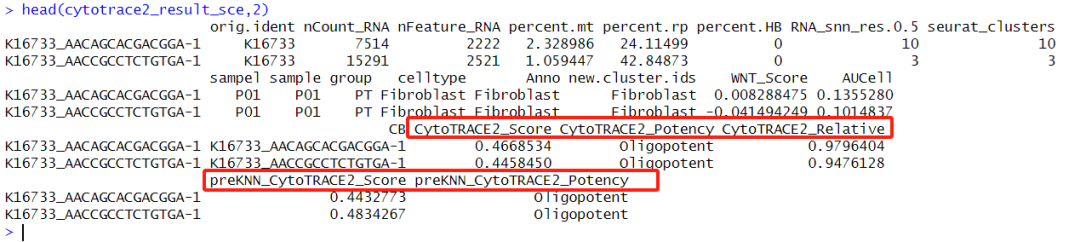
Τδ÷–CytoTRACE2_RelativeΈΣscoreΒΡΨΏΧε ΐ÷ΒΫαΙϊΘΜCytoTRACE2_PotencyΈΣΈΡ’¬ΩΣΆΖΧαΒΫΒΡΒΡΝυάύΫαΙϊΓΘ
ΉΔ1ΘΚcytotrace2Ρ§»œΒΡ «–Γ σΘ§Υυ“‘–η“Σ÷ΗΕ®species = 'human' ΘΜ»γΙϊ «ΒΞœΗΑϊΕ‘œσΒΡΜΑ–η“Σ÷ΗΕ®is_seurat = TRUE ΘΜ÷ΗΕ®seed ΖΫ±ψΚσ–χΒΡΫαΙϊΗ¥œ÷ΓΘΓΘ
2Θ§CytoTRACE v2Ω… ”Μ·
Θ®1Θ©v2‘Ύ plotData
Ά§cytotrace v1ΒΡΩ… ”Μ·Κ· ΐ≤Μ“Μ―υΘ§v2‘Ύ plotDataΚ· ΐ÷–ΑϋΉΑΝΥ“Μ–©≥ΘΦϊΒΡΩ… ”Μ·ΫαΙϊ Θ§Ω…“‘œ»…ηΕ®¥ΐ’Ι ΨΒΡ±μ–ΆΘ®celltypeΘ© ΓΘ
# making an annotation dataframe that matches input requirements for plotData function
annotation <- data.frame(phenotype = sce3@meta.data$celltype) %>%
set_rownames(., colnames(sce3))
# plotting
plots <- plotData(cytotrace2_result = cytotrace2_result_sce,
annotation = annotation,
is_seurat = TRUE)
# Μφ÷ΤCytoTRACE2_PotencyΒΡumapΆΦ
p1 <- plots$CytoTRACE2_UMAP
# Μφ÷ΤCytoTRACE2_PotencyΒΡumapΆΦ
p2 <- plots$CytoTRACE2_Potency_UMAP
# Μφ÷ΤCytoTRACE2_RelativeΒΡumapΆΦ Θ§v1
p3 <- plots$CytoTRACE2_Relative_UMAP
# Μφ÷ΤΗςœΗΑϊάύ–ΆCytoTRACE2_ScoreΒΡœδœΏΆΦ
p4 <- plots$CytoTRACE2_Boxplot_byPheno
(p1+p2+p3+p4) + plot_layout(ncol = 2)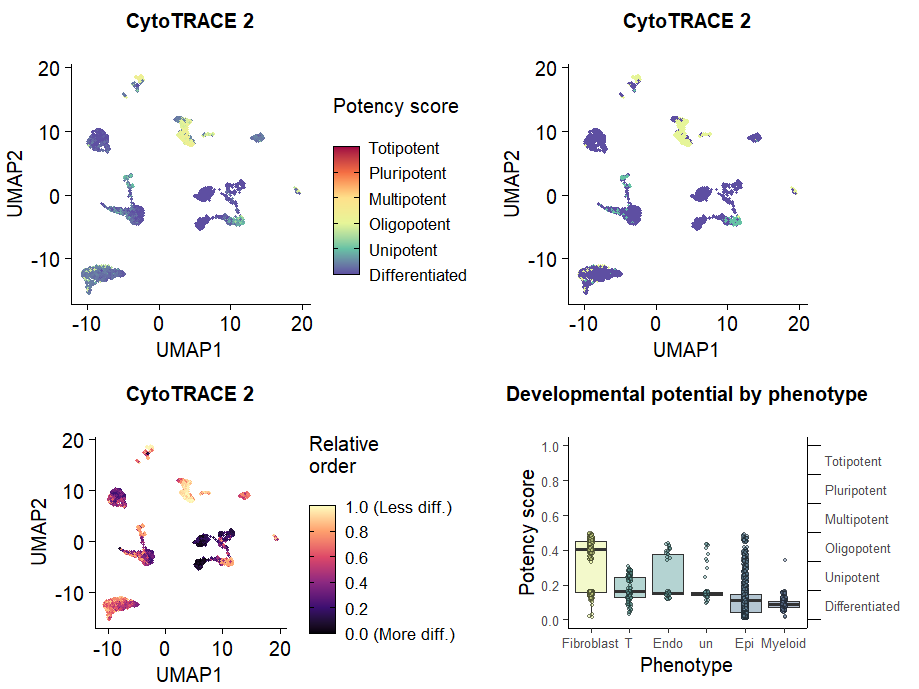
Θ®2Θ©Βς’ϊ≥ωΆΦΒΡΖγΗώΘ§”κV1Ϋ”ΫϋΘ®plotDataΚ· ΐ÷–ΒΡ¥ζ¬κΘ©
FeaturePlot(cytotrace2_result_sce, "CytoTRACE2_Relative",pt.size = 1.5) +
scale_colour_gradientn(colours =
(c("#9E0142", "#F46D43", "#FEE08B", "#E6F598",
"#66C2A5", "#5E4FA2")),
na.value = "transparent",
limits = c(0, 1),
breaks = seq(0, 1, by = 0.2),
labels = c("0.0 (More diff.)",
"0.2", "0.4", "0.6", "0.8", "1.0 (Less diff.)"),
name = "Relative\norder \n",
guide = guide_colorbar(frame.colour = "black",
ticks.colour = "black")) +
ggtitle("CytoTRACE 2") +
xlab("UMAP1") + ylab("UMAP2") +
theme(legend.text = element_text(size = 10),
legend.title = element_text(size = 12),
axis.text = element_text(size = 12),
axis.title = element_text(size = 12),
plot.title = element_text(size = 12,
face = "bold", hjust = 0.5,
margin = margin(b = 20))) +
theme(aspect.ratio = 1)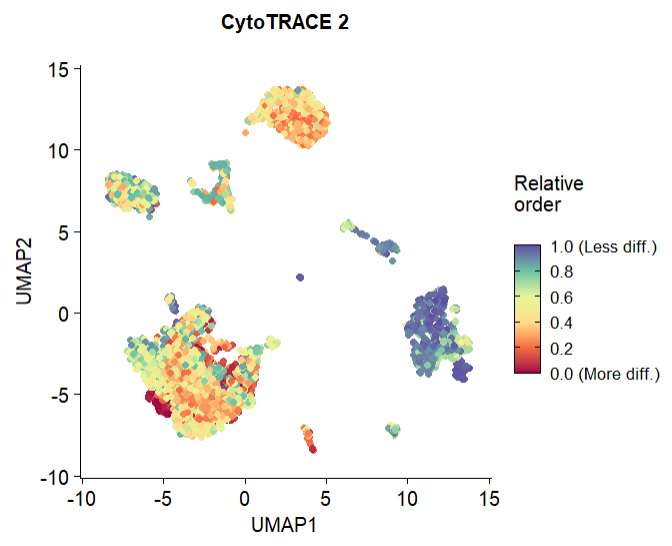
ΒΞœΗΑϊΒΡΚήΕύΩ… ”Μ·ΕΦ «Ω…“‘ Ι”Οggplot2Ϋχ––Ή‘Ε®“εΒΡΓΘΗϋΕύggplot2 ΒΡΒς’ϊΩ…“‘≤ΈΩΦggplot2 | ΙΊ”Ύ±ξΧβΘ§Ήχ±ξ÷αΚΆΆΦάΐΒΡœΗΫΎ–όΗΡΘ§ΡψΩ…ΡήœκΝΥΫβ,ggplot2|œξΫβΑΥ¥σΜυ±ΨΜφΆΦ“ΣΥΊ,ggplot2|theme÷ςΧβ…η÷ΟΘ§œξΫβΜφΆΦ”≈Μ·-ΓΑΨΪΒώœΗΉΝΓ± Β» ΓΘ
Θ®3Θ©œΗΑϊάύ–Ά-œδœΏΆΦ
≥ΐΝΥp4Ή‘¥χΒΡœδœΏΆΦΘ§“≤Ω…“‘ΗυΨί–η«σΉ‘––Μφ÷Τ scRNAΖ÷Έω| Ι”ΟAddModuleScore ΚΆ AUcellΫχ––Μυ“ρΦ·¥ρΖ÷Θ§Ω… ”Μ·
library(ggpubr)
p1 <- ggboxplot(cytotrace2_result_sce@meta.data, x="celltype", y="CytoTRACE2_Score", width = 0.6,
color = "black",#¬÷άΣ―’…Ϊ
fill="celltype",#Χν≥δ
palette = "npg",
xlab = F, #≤Μœ‘ Ψx÷αΒΡ±ξ«©
bxp.errorbar=T,#œ‘ ΨΈσ≤νΧθ
bxp.errorbar.width=0.5, #Έσ≤νΧθ¥σ–Γ
size=1, #œδ–ΆΆΦ±ΏœΏΒΡ¥÷œΗ
outlier.shape=NA, #≤Μœ‘ Ψoutlier
legend = "right") #ΆΦάΐΖ≈”“±Ώ
###÷ΗΕ®Ήι±»Ϋœ
my_comparisons <- list(c("Epi", "un"), c("T", "un"),c("Myeloid", "un"))
p1+stat_compare_means(comparisons = my_comparisons,
method = "wilcox.test")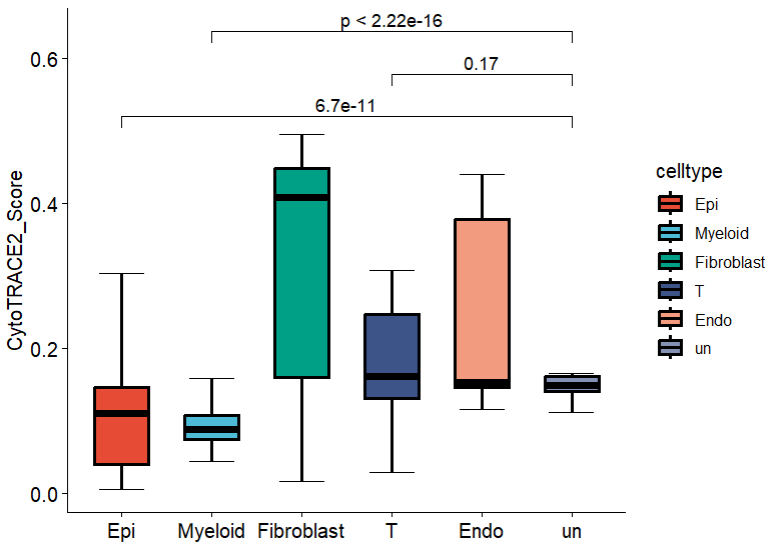
3Θ§ΫαΚœmonocle2 »ΖΕ®ΤπΒψ
œύΙΊΒΡ‘Λ≤βΫαΙϊ“―Ψ≠‘Ύmetadata÷–ΝΥΘ§Ω…“‘‘Ύmonocle2÷–Μφ÷ΤΜυ”ΎΖ÷Μ· scoreΒΡΫαΙϊΘ§“‘¥Υά¥Αο÷ζ»ΖΕ®ΤπΒψΓΘ
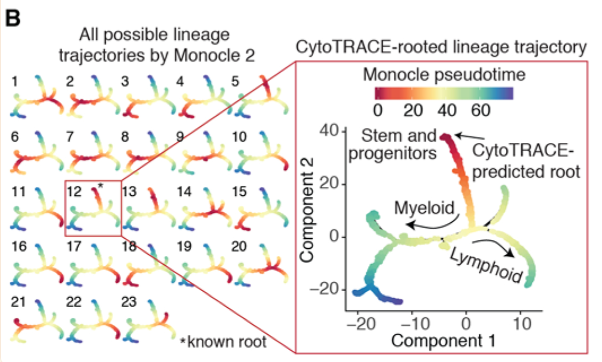
≤ΈΩΦΉ ΝœΘΚ
[1]Mapping single-cell developmental potential in health and disease with interpretable deep learning
[2]Single-cell transcriptional diversity is a hallmark of developmental potential
±ΨΈΡΖ÷œμΉ‘ …ζ–≈≤ΙΗχ’Ψ ΈΔ–≈ΙΪ÷ΎΚ≈Θ§«ΑΆυ≤ιΩ¥
»γ”–«÷»®Θ§«κΝΣœΒ cloudcommunity@tencent.com …Ψ≥ΐΓΘ
±ΨΈΡ≤Έ”κ?ΧΎ―Ε‘ΤΉ‘ΟΫΧεΖ÷œμΦΤΜ°? Θ§ΜΕ”≠»»Α°–¥ΉςΒΡΡψ“ΜΤπ≤Έ”κΘΓ