Nat. Methods | 一个用于预测动态环境中全新蛋白-蛋白相互作用的平台
Nat. Methods | 一个用于预测动态环境中全新蛋白-蛋白相互作用的平台

今天为大家介绍的是来自Ileana. M. Cristea团队的一篇论文。蛋白-蛋白相互作用(PPIs)推动细胞过程和对环境的反应,反映了细胞状态。在这里,作者开发了Tapioca,一个用于研究动态环境中全局PPIs的集成机器学习框架。

在测定动态蛋白-蛋白相互作用时的一个挑战是,这些复杂数据集,包括准确识别全新PPIs的需求,尚未得到通用、准确和稳健的计算及分析框架的满足。在TPCA和I-PISA背景下准确预测PPIs的困难在于,许多蛋白质与它们不相互作用的其他蛋白质的曲线数据非常接近。避免高假阳性率(FPR)需要严格的距离截断,导致许多真实相互作用的漏检。在这里,作者开发了Tapioca,一个集成的基于机器学习的计算管道,用于全新预测动态PPIs。Tapioca整合了来自TPCA(Thermal denaturation)、CF-MS(cofractionation- mass spectrometry)或I-PISA(Ion-based precipitation)的MS曲线数据,以及蛋白质属性和组织特异性功能网络。与基于欧几里得距离的方法相比,Tapioca具有更优的预测性能。通过生成Tapioca获得的洞见,作者改进了TPCA的实验工作流程,以提高PPI预测的效率。作者使用计算机模拟和实验方法优化了热变性的温度范围,减少了温度点的数量并提高了吞吐量,以及改善了细胞裂解条件,增加了检测到的蛋白质(定位)的数量和多样性,以及PPI预测的质量。
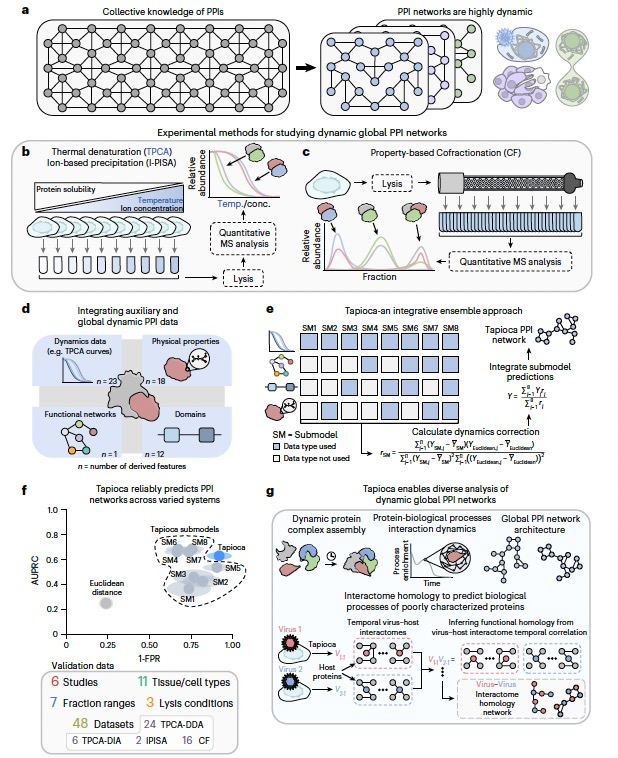
图 1
动态环境计算PPI
为了满足在全局范围内确定蛋白-蛋白相互作用(PPIs)动态的计算工具需求,作者开发了Tapioca,一个基于逻辑回归的集成机器学习框架。Tapioca提供了一种手段,可以将来自TPCA、I-PISA或CF-MS数据的基于曲线的动态PPI数据与静态交互数据整合起来,准确地预测动态环境中的PPIs。Tapioca利用的先前交互知识包括序列预测的蛋白质物理属性、来自Pfam数据库的领域信息以及组织特异性功能网络(图1d)。这些功能网络是基于来自成千上万个集成的“-omics”数据集(例如,基因共表达、转录因子结合和蛋白质-蛋白质相互作用)的生物学信息,使用贝叶斯概率框架构建的。贝叶斯机器学习模型自动增加了对感兴趣组织信息量大的数据集的权重,从而在整合中实现了组织特异性。通过识别紧密连接的共享功能关联的蛋白质,尽管这些关联不一定通过直接的PPIs,这些功能网络提供了在多样化细胞途径中预测蛋白质行为的地图。
Tapioca由八个子模型组成,每个子模型都利用来自质谱分析的静态交互和动态数据的独特组合进行预测(图1e)。每个子模型都对给定数据集(例如,TPCA数据集)中存在的PPIs进行预测。这些子模型得分的整合被优化,以通过计算动态修正得分(图1e)来获得PPI预测的准确性与系统动态性的平衡。Tapioca在的六个TPCA数据集上进行了训练,并使用48个独立数据集进行评估,这些数据集包括30个TPCA、16个CF和2个I-PISA数据集,代表了11种组织/细胞类型,使用五折交叉验证的标准蛋白质相互作用进行。在研究中进一步分析的Tapioca的最终版本,是在70%训练集,30%测试集的划分上训练和测试的(图1f)。作者使用不同的机器学习算法评估了Tapioca工作流程——逻辑回归、朴素贝叶斯和随机森林。作者发现逻辑回归在PPI预测的准确性和系统动态性捕捉之间提供了适当的平衡。Tapioca在预测PPIs方面大大优于欧几里得距离(图1f),这是从TPCA或I-PISA数据预测PPIs的传统方法,并且在广泛的实验方法和生物学环境中表现良好。
提高预测的健壮性

图 2
Tapioca使用一组在各种实验环境中观察到的、定义明确的CORUM复合体作为黄金标准进行训练。作者使用这个静态黄金标准是因为没有一个经过实验验证的全球PPI动态数据集足够大且具有足够多样化的生物学环境,可以用作替代的动态黄金标准。静态黄金标准的一个缺点是,它可能会降低训练于此的子模型捕捉系统动态的能力。这是因为子模型使用了静态交互数据,可能会更多地依赖与先前知识(静态数据)相关的特征,实际上减少了动态数据(例如,TPCA、I-PISA或CF数据)对子模型预测的贡献。为了理解增加子模型使用的静态交互数据量对其捕捉系统动态能力的影响,作者比较了纤维细胞在未感染和感染HSV-1后的预测互作组。HSV-1感染会引起宿主细胞蛋白质组和PPI网络的全局变化,且在感染后15小时(即感染的后期阶段)的蛋白质互作组应该反映这种变化(图2a)。虽然PPI预测质量随着子模型使用的先前知识量增加而普遍提高,但其捕捉系统动态的能力却减少了(图2b)。这表明,正如预期的那样,子模型在进行预测时通常会过分依赖静态交互数据,从而低估了系统动态。Tapioca通过将这些子模型与动态数据整合,平衡了准确的PPI预测与捕获系统中的生物扰动。
作者进一步比较了Tapioca整合子模型的方法与其他方法(图2c)。从简单的整合方法开始,例如使用子模型预测的平均值(均值、中位数)或它们在黄金标准上的表现(曲线下面积(AUC)、精确度-召回曲线下面积(AUPRC)),确实观察到了系统动态的一些预测(图2d)。然而,这些方法是静态问题的静态解决方案,无论数据如何(均值、中位数),都以相同的方式结合预测,或者是根据它们满足静态黄金标准的期望程度进行组合(AUC、AUPRC)。因此,作者提出了一个动态驱动的解决方案,取子模型得分与基于欧几里得距离导出的得分的皮尔森相关性。这种相关性被用于加权整合子模型得分。Tapioca采用这种方法进行子模型整合,提供了最佳性能,平衡了系统动态与可靠的PPI预测(图2d)。
Taploca泛化性

图 3
Tapioca旨在成为一个泛化的工具,用于从基于曲线的动态数据预测PPIs,其特征生成对曲线的分辨率或形状不敏感。最初在TPCA数据集上训练的Tapioca,能够准确地从I-PISA数据预测PPIs,而且更重要的是,尽管TPCA和CF数据之间的曲线形状有很大差异,也能从CF数据准确预测PPIs。为了进一步测试Tapioca的泛化能力,作者评估了它在异质动态数据(HDD)上的性能,这些数据是通过组合已发布的CF和TPCA数据集(CF-TPCA)或I-PISA和TPCA数据集(I-PISA-TPCA)从相似(CF-TPCA38,40)或相同(I-PISA-TPCA22)的生物学环境生成的(图3a)。HDD数据集的表现通常与它们的父数据集相当或更好(图3b)。Tapioca通常会给予来自HDD的相互作用更保守的评分,因此给出的PPI预测较少(图3c)。因此,尽管可能会丢失某些特定由一个实验方法特有捕获的相互作用,整合异质数据集有助于突出高置信度PPI预测的子集。为了更好地理解使用HDD或父数据集捕获的PPI网络,作者评估了这些数据集中以相似置信度识别的共有和独特相互作用。正如预期的那样,来自HDD分析的评分与来自父源的评分显示出适度相关(图3d),表明从HDD数据集也可以获得独特的PPI预测。在考虑总的PPI预测时,与它们的父数据集相比,HDD数据集提供了中等数量的相互作用(图3e)。
编译 | 曾全晨
审稿 | 王建民
参考资料
Reed, T.J., Tyl, M.D., Tadych, A. et al. Tapioca: a platform for predicting de novo protein–protein interactions in dynamic contexts. Nat Methods 21, 488–500 (2024).
https://doi.org/10.1038/s41592-024-02179-9