ncount_RNA 和nFeature_RNA辅助过滤
ncount_RNA 和nFeature_RNA辅助过滤

前情提要
上次给大家简单整理了一下细胞鉴定曲线图理解,里面使用nCount_RNA或者nFeature_RNA在R语言里面绘制细胞鉴定曲线,找到一个合适的cutoff值,进行了一个初步的质控。
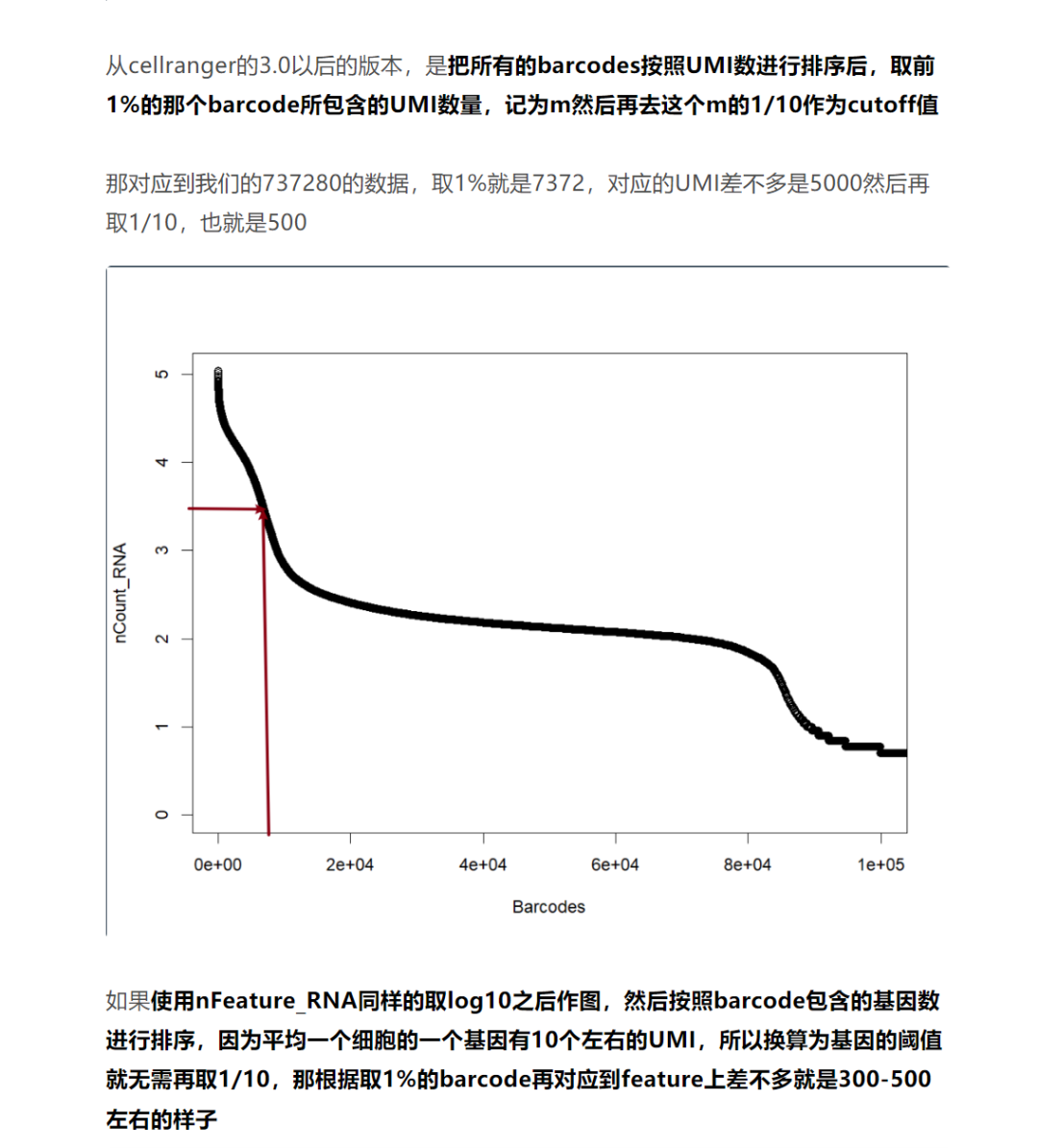
结尾也提到了,很少会有下游是原始的rawcounts的数据,一般我们都是使用cellranger质控后的数据进行分析。不过对于处理后的数据集我们可以可视化一下nFeature_RNA和nCount_RNA来辅助进行质控
那首先我们基于Seurat官网的教程来了解回顾一下nFeature_RNA和nCount_RNA,并且可视化判断一下阈值,然后了解一下实际分析情况中的应用。
nFeature_RNA和nCount_RNA简介
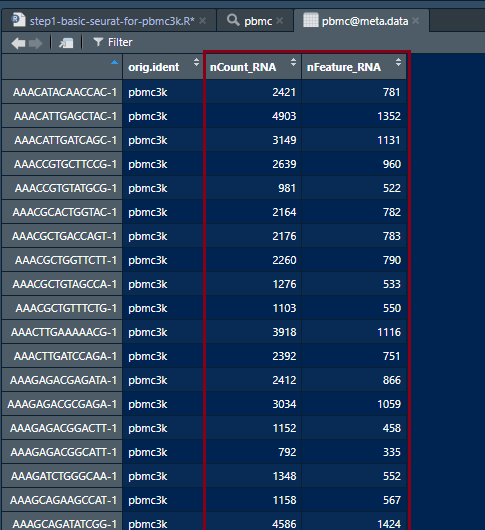
创建完seurat对象之后,在不进行任何操作时,seurat会为每个细胞创建一个元数据,保存在meta.data里面
#读取数据创建seurat对象
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data,
project = "pbmc3k",
min.cells = 3)
> dim(pbmc)
[1] 13714 2700

每一列的内容:
orig.ident:通常包含所知的样品名,默认为我们赋给project的值,如果不赋值那就是SeuratProjectnCount_RNA:每个细胞的UMI数目nFeature_RNA:每个细胞所检测到的基因数目
可以看到nCount_RNA和nFeature_RNA还是有差异的,这就与它们的计算方法有关
#nCount_RNA:总的UMI数即转录本数量
colSums(sce@assays$RNA$counts)
#nFeature_RNA:总的基因数目
colSums(sce@assays$RNA$counts>0)
可视化及阈值判断
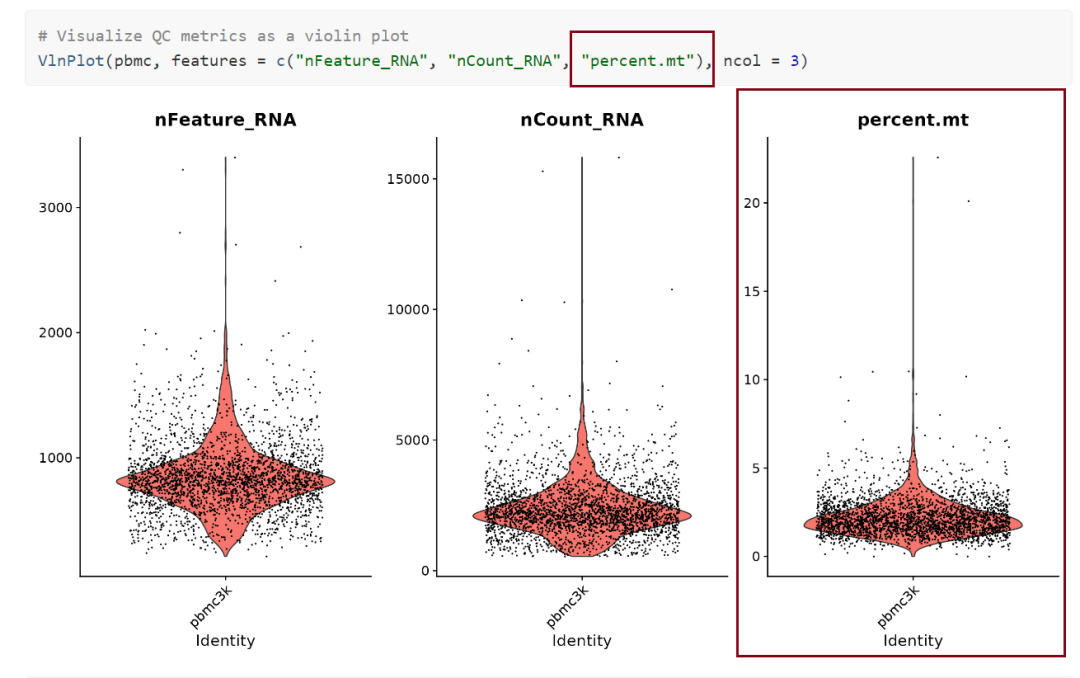
可以使用小提琴图来简单可视化一下nFeature_RNA和nCount_RNA
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA"))
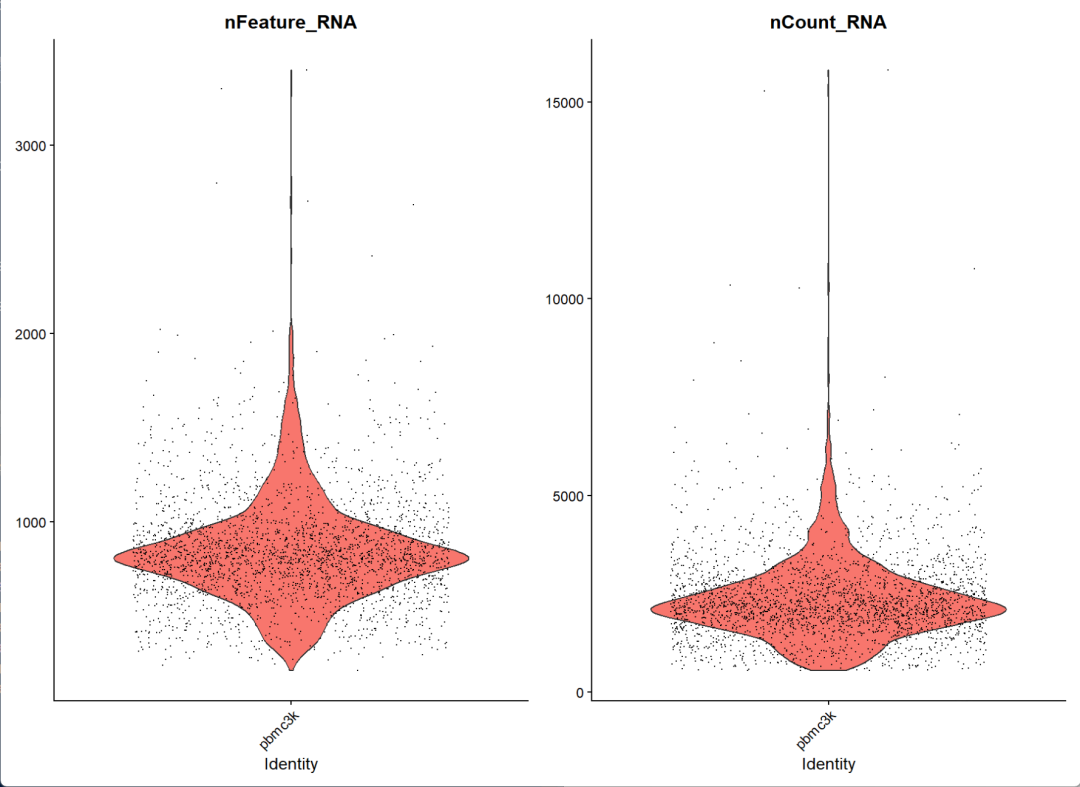
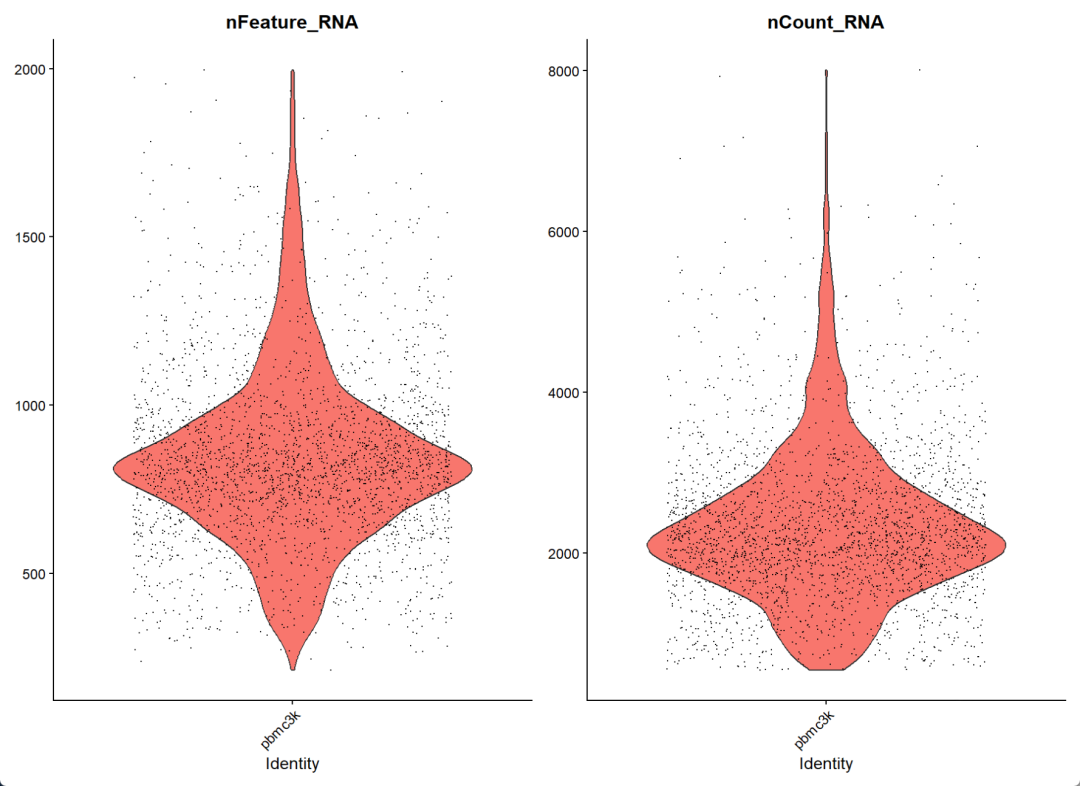
过滤前
nFeature_RNA图:反映的是样品中每个细胞表达的基因数量,表达过高可能是双细胞或者多细胞,表达过低可能是空液滴或者包裹的是环境RNA
nCount_RNA图:反映的是每个细胞中包含的UMI数量也就是转录本的数量
在10X Genomics测序数据分析过程中,通过UMI对测序得到的reads进行简并之后,就可以看到一个细胞中被读到多少个基因。一般一个细胞可以得到40000-80000个有效的UMI,平均一个细胞的一个基因有10个左右的UMI。
所以我们在进行阈值判断的时候,可以直接基于nFeature_RNA值也就是基因的数量
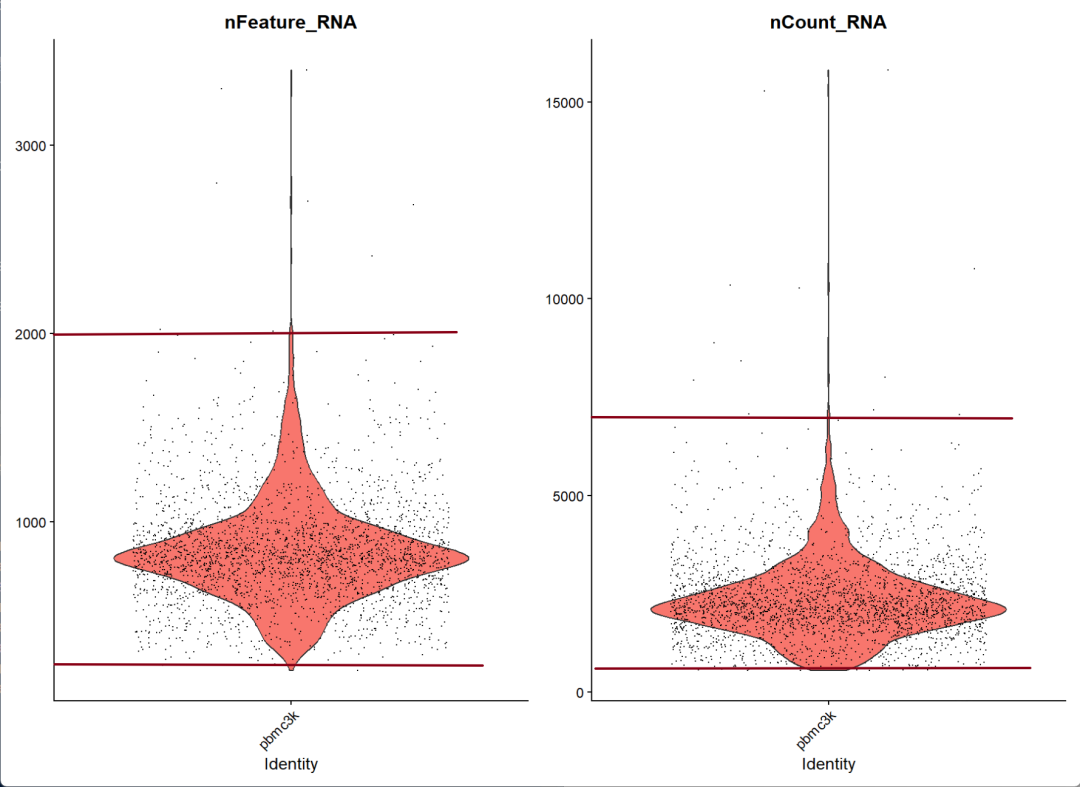
阈值判断
We filter cells that have unique feature counts over 2,500 or less than 200
官网给的是大于200和小于2500,但可视化之后我们可以看到上限定在2000其实也可以。
不过pbmc是比较早期的数据了,测到的细胞数量比较少,上限设置的也比较低,如果是现在的单细胞数据还是要具体数据具体分析
#基于阈值过滤并且可视化
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2000)
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA"))
> dim(pbmc)
[1] 13714 2692
过滤后
过滤后,细胞从最开始的2700变为现在的2692,过滤掉了部分细胞。
以上是seurat官网pbmc3k_tutorial中QC的部分内容,接下来我们看看在实际数据中的应用。
实际分析中应用
如果大家手里有技能树单细胞分析的标准分析代码,如果需要的话可以获取一下链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh
那在我们的scRNA_scripts文件夹中有个qc.R的质控脚本文件,就是对读取进来的数据进行质控的。
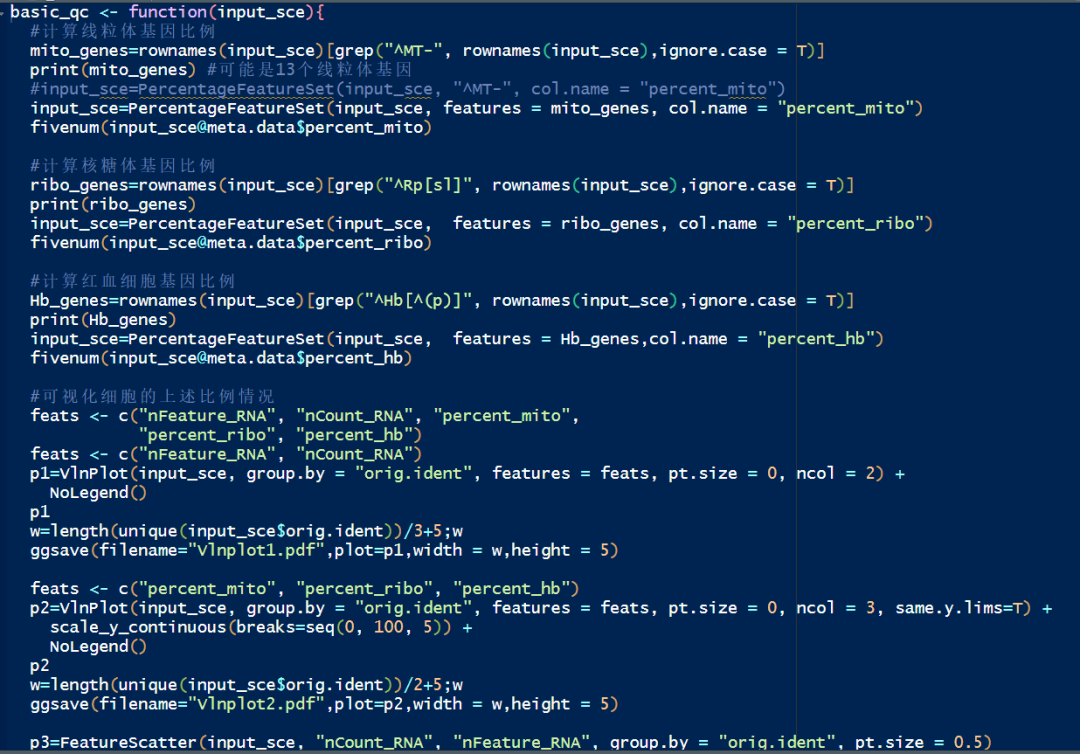
脚本函数首先是计算了线粒体、核糖体以及血红细胞的比例(下期给大家详细介绍),然后就可视化了细胞中这些参数的情况。我们还是先重点看看nFeature_RNA和nCount_RNA
#qc.R脚本中nFeature_RNA和nCount_RNA部分内容
feats <- c("nFeature_RNA", "nCount_RNA")
p1=VlnPlot(input_sce, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
p1
w=length(unique(input_sce$orig.ident))/3+5;w
ggsave(filename="Vlnplot1.pdf",plot=p1,width = w,height = 5)

质控前
一般走标准流程的时候,在创建seurat对象时候就会基于min.cells = 5和min.features = 300进行过滤,所以在qc脚本中是不进行这一步的过滤操作的。不过为了看一下过滤前后变化,我们可以基于可视化的结果进行一个简单的过滤操作。

#简单过滤
if(T){
selected_c <- WhichCells(input_sce, expression = nFeature_RNA > 500 & nFeature_RNA < 2500)
selected_f <- rownames(input_sce)[Matrix::rowSums(input_sce@assays$RNA$counts > 0 ) > 3]
input_sce.filt <- subset(input_sce, features = selected_f, cells = selected_c)
dim(input_sce)
dim(input_sce.filt)
}
#可视化过滤后的情况
feats <- c("nFeature_RNA", "nCount_RNA")
p1_filtered=VlnPlot(input_sce.filt, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 2) +
NoLegend()
w=length(unique(input_sce.filt$orig.ident))/3+5;w
ggsave(filename="Vlnplot1_filtered.pdf",plot=p1_filtered,width = w,height = 5)
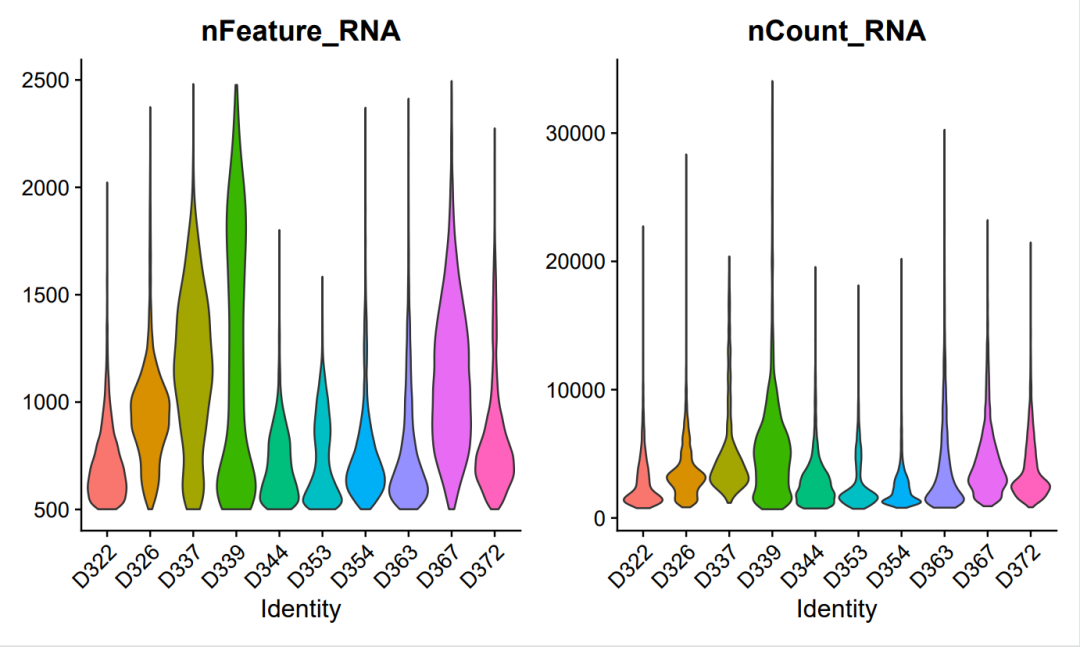
过滤后
基本质控意义:可以去除掉每个样品中,一些表达量过高或者过低的基因。
除了在基本质控环节我们会可视化一下细胞中nFeature_RNA和nCount_RNA的情况,在进行降维聚类分群的时候,我们也会对nFeature_RNA和nCount_RNA进行可视化。
细胞降维聚类分群中应用
在选择对应的阈值进行可视化的时候,我们会用到check-all-markers.R脚本,基于常见Marker基因进行一下可视化,以及绘制umap图

在check-all-markers.R脚本,帮助我们查看确认每个细胞亚群中基因的表达情况,从而帮助我们判断是否是双细胞。
具体推文:如何排除双细胞
我们在进行亚群简单命名的时候,一般选择比较低的分辨率0.1,那在GSE208706数据的0.1分群中,我们可以很明显的看到第9群比较狭长,且包含了两个不同细胞亚群的Marker基因。
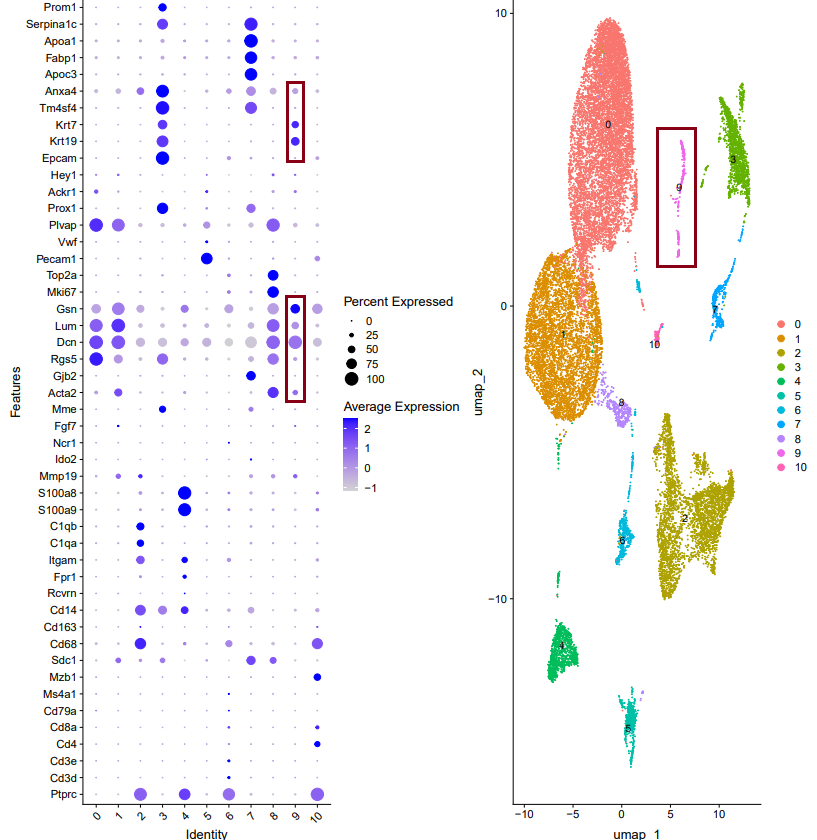
为了判断是否是双细胞,我们就需要结合每个亚群的单个细胞的总的RNA数量进行判断
if("percent_mito" %in% colnames(sce.all.int@meta.data ) ){
#可视化细胞的上述比例情况
feats <- c("nFeature_RNA", "nCount_RNA", "percent_mito", "percent_ribo", "percent_hb")
feats <- c("nFeature_RNA", "nCount_RNA")
p1=VlnPlot(sce.all.int , features = feats, pt.size = 0, ncol = 2) +
NoLegend()
w=length(unique(sce.all.int$orig.ident))/3+5;w
ggsave(filename=paste0(pro,"Vlnplot1.pdf"),plot=p1,width = w,height = 5)
feats <- c("percent_mito", "percent_ribo", "percent_hb")
p2=VlnPlot(sce.all.int, features = feats, pt.size = 0, ncol = 3, same.y.lims=T) +
scale_y_continuous(breaks=seq(0, 100, 5)) +
NoLegend()
w=length(unique(sce.all.int$orig.ident))/2+5;w
ggsave(filename=paste0(pro,"Vlnplot2.pdf"),plot=p2,width = w,height = 5)
}

nFeature_RNA可视化结果发现反而第8群表达量高,而第9群正常。基于Marker基因推断第8群是处于增殖期的细胞,所以表达量高是合理的。
并且提高分辨率之后,发现9群被细分为两个亚群,也不是双细胞。
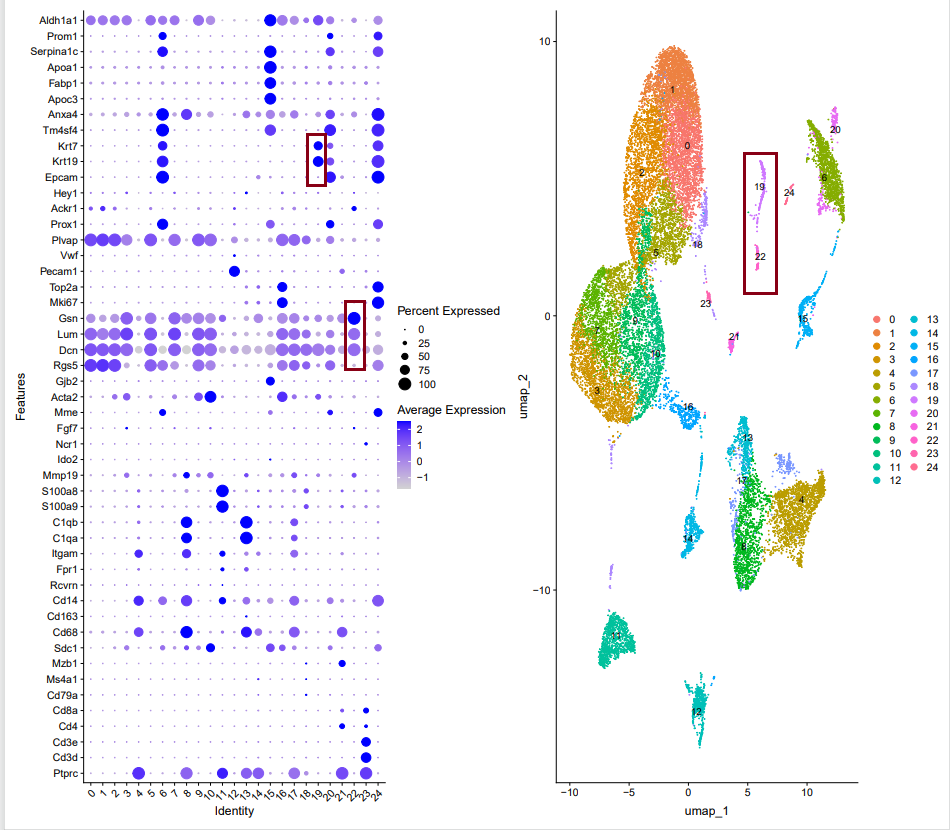
一般我们会根据中位线以及最高值来进行判断,再提高分辨率看亚群有没有分开,再确定是否是双细胞。
线粒体比例
在官网以及我们的标准质控流程中,都会计算线粒体比例
我们的qc.R脚本中还对核糖体以及血红细胞的比例进行了计算和可视化,那下期一起来了解一下这些内容吧!
