根据线粒体基因进行过滤

前情提要
上篇推文中对ncount_RNA 和nFeature_RNA进行了可视化,然后基于可视化结果进行了阈值的判断,并且也给大家分享了在实际分析中的应用
其中也提到了在我们的质控脚本中,首先是计算了线粒体、核糖体以及血红细胞的比例,然后就可视化了细胞中这些参数的情况,在基于这些数据进行一个过滤
那这期我们来了解一下如何根据线粒体、核糖体以及红血蛋白基因的比例,对细胞进行过滤
为什么要基于这些基因进行过滤
单细胞中表达量最高的基因一般是线粒体基因、核糖体基因等
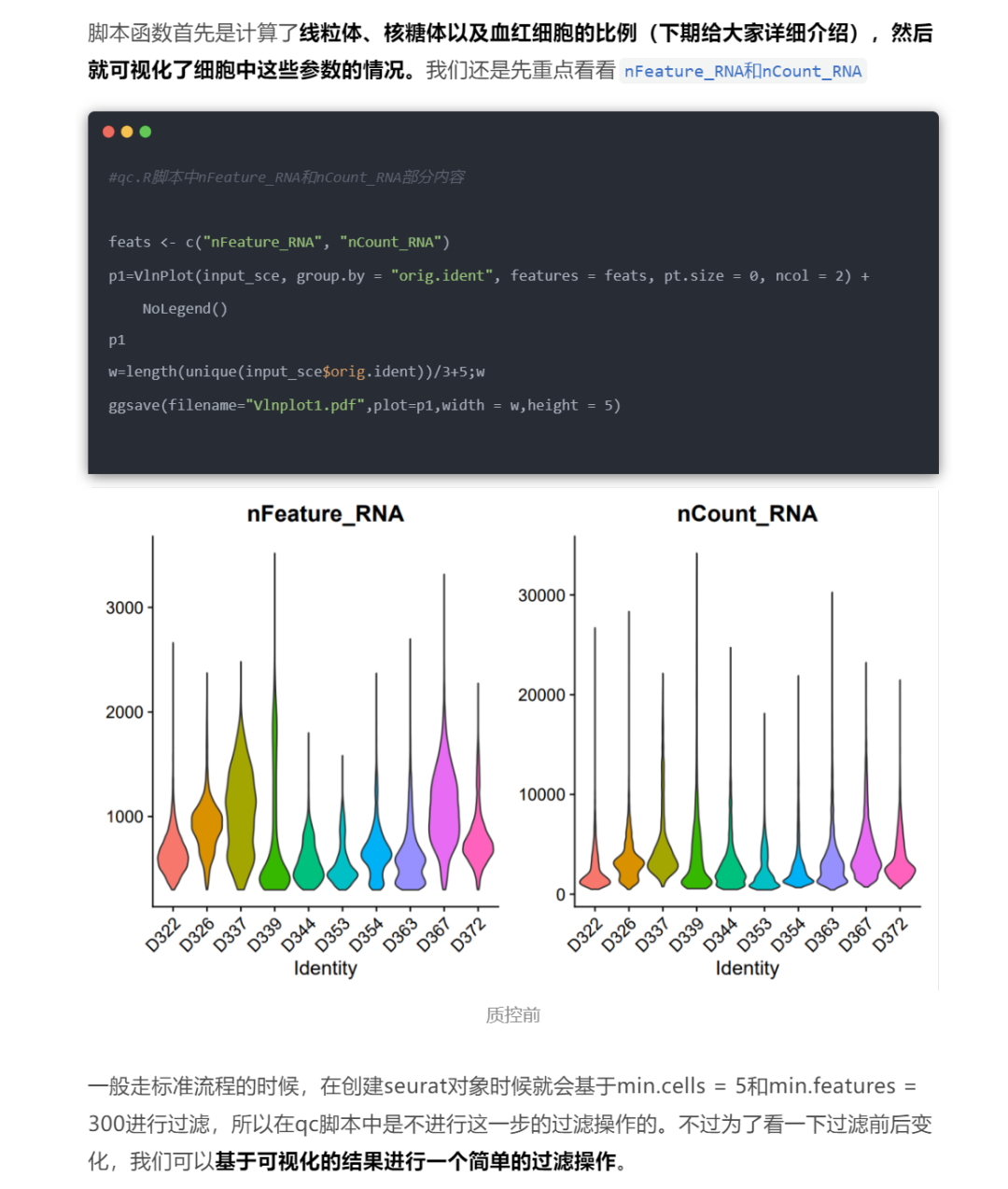
#抽样查看TOP50基因
# 这里的C 这个矩阵,有一点大,可以考虑随抽样
C=subset(sce.all,downsample=1000)@assays$RNA$counts
dim(C)
dim(sce.all)
C=Matrix::t(Matrix::t(C)/Matrix::colSums(C)) * 100
most_expressed <- order(apply(C, 1, median), decreasing = T)[50:1]
pdf("TOP50_most_expressed_gene.pdf",width=14)
pdf("test.pdf",width = 15,height = 15)
boxplot(as.matrix(Matrix::t(C[most_expressed, ])),
cex = 0.2, las = 2,
ylim=c(0,8),
xlab = "% total count per cell",
col = (scales::hue_pal())(50)[50:1],
horizontal = TRUE)
dev.off()
如果是线粒体基因高表达的细胞,可能是处于凋亡状态或者裂解状态的细胞,会干扰后续的分析
红细胞中会高表达血红蛋白基因,而红细胞本身是不含有细胞核的,且携带的基因少,其携带的基因与疾病以及生物发育等过程没有太大的联系,所以可以直接剔除掉
计算基因比例
在读取完数据创建seurat对象之后,会为每个细胞创建一个元数据,保存在meta.data里面,比如我们上次介绍的nFeature_RNA和nCount_RNA,统计一下全部基因的表达量
但是并不会计算线粒体、核糖体这些单独的基因的比例,所以需要我们自行计算一下这些基因,然后也保存在meta.data里面
计算方法:
- 根据基因名特征进行整理
- 使用
PercentageFeatureSet进行计算
#计算线粒体基因比例
mito_genes=rownames(sce.all)[grep("^MT-", rownames(sce.all),ignore.case = T)]
print(mito_genes)
sce.all=PercentageFeatureSet(sce.all, features = mito_genes, col.name = "percent_mito")
fivenum(sce.all@meta.data$percent_mito)
#计算核糖体基因比例
ribo_genes=rownames(sce.all)[grep("^Rp[sl]", rownames(sce.all),ignore.case = T)]
print(ribo_genes)
sce.all=PercentageFeatureSet(sce.all, features = ribo_genes, col.name = "percent_ribo")
fivenum(sce.all@meta.data$percent_ribo)
#计算红血细胞基因比例
Hb_genes=rownames(sce.all)[grep("^Hb[^(p)]", rownames(sce.all),ignore.case = T)]
print(Hb_genes)
sce.all=PercentageFeatureSet(sce.all, features = Hb_genes,col.name = "percent_hb")
fivenum(sce.all@meta.data$percent_hb)

人类和小鼠的线粒体核糖体基因区别:
- 人类的核糖体基因是RPS/RPL开头
- 人类的线粒体基因是MT-开头
- 小鼠的核糖体基因是Rps/Rpl开头
- 小鼠的线粒体基因是mt-开头
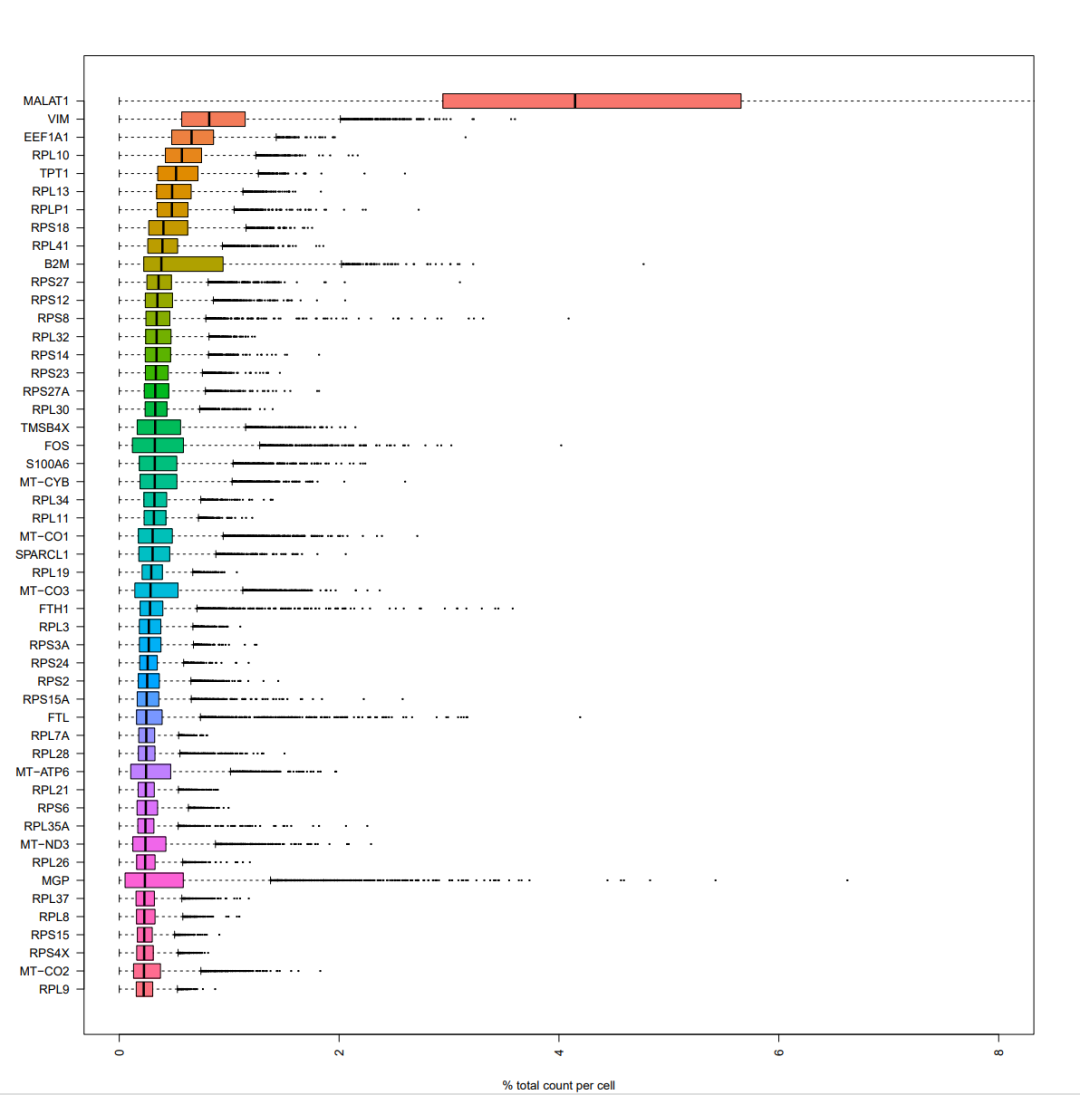
可视化
feats <- c("percent_mito", "percent_ribo", "percent_hb")1
p2=VlnPlot(sce.all, group.by = "orig.ident", features = feats, pt.size = 0, ncol = 3, same.y.lims=T) +
scale_y_continuous(breaks=seq(0, 100, 5)) +
NoLegend()
p2
如果分析中发现某些单细胞样品中的线粒体表达量特别高,可能说明这个样品质量是比较一般的
设置阈值过滤
一般简单的过滤就是基于可视化的结果,设置一个上限
#过滤指标2:线粒体/核糖体基因比例(根据上面的violin图)
selected_mito <- WhichCells(sce.all, expression = percent_mito < 25)
#selected_ribo <- WhichCells(sce.all, expression = percent_ribo > 3)
selected_hb <- WhichCells(sce.all, expression = percent_hb < 1 )
length(selected_hb)
length(selected_ribo)
length(selected_mito)
sce.all_filt <- subset(sce.all, cells = selected_mito)
#sce.all <- subset(sce.all, cells = selected_ribo)
sce.all_filt <- subset(sce.all_filt, cells = selected_hb)
dim(sce.all_filt)
table(sce.all_filt$orig.ident)

根据线粒体核糖体基因进行过滤
在过滤线粒体核糖体基因推文中提到了过滤的方式
1. 粗暴的去除全部线粒体基因
直接在表达矩阵里面,去除掉属于的那一行表达量即可
2. 设定阈值去除掉高表达量细胞
多次反复查看线粒体核糖体基因的影响,分群前后看,不同batch看,多次质控图表里面显示它,判断它是否是一个主要因素
3. 高表达核糖体基因的细胞是否要去除
在不影响分群和功能分析时可以不删除,很多细胞里都大量表达核糖体基因,如果一刀切全删除,会删除很多目的细胞。
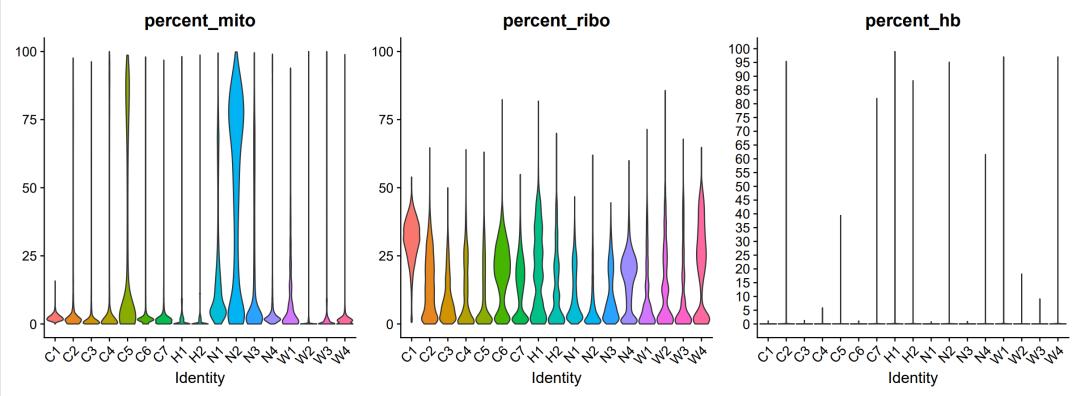
在分析中的应用
与ncount_RNA 和nFeature_RNA一样,在进行单细胞亚群可视化的时候,也会可视化一下线粒体核糖体基因这些基因的比例
feats <- c("percent_mito", "percent_ribo", "percent_hb")
p2=VlnPlot(sce.all.int, features = feats, pt.size = 0, ncol = 3, same.y.lims=T) +
scale_y_continuous(breaks=seq(0, 100, 5)) +
NoLegend()
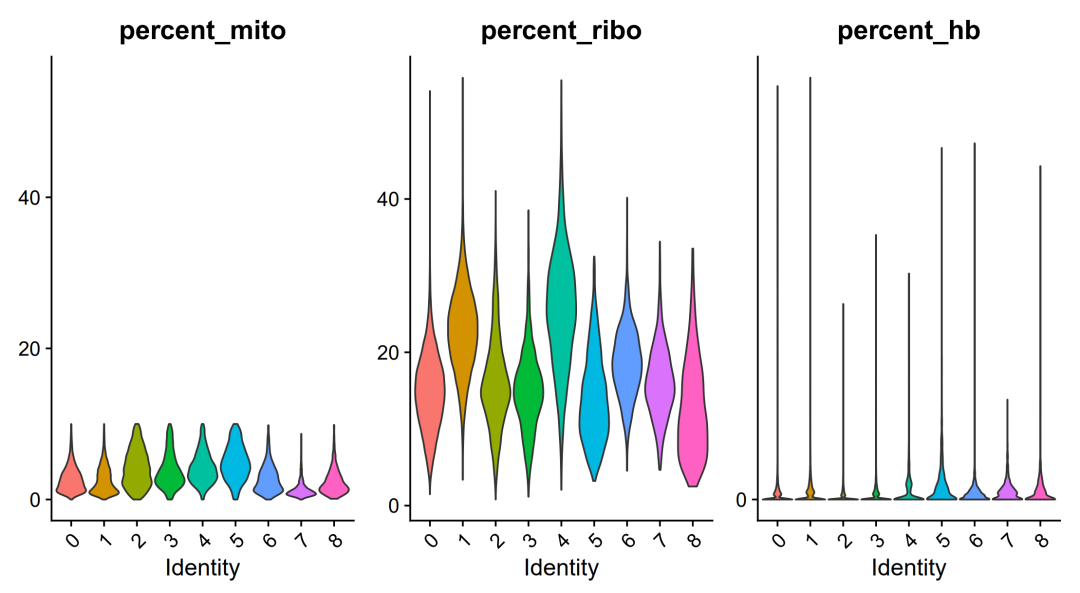
不过因为在质控的时候已经去除掉线粒体基因表达过高的细胞,所以在亚群分类的时候也不太需要仔细辨别
在单细胞亚群细分的时候,主要还是基于每个簇的ncount_RNA 和nFeature_RNA以及基于Marker基因和Umap图去判断是否是双细胞,或者增殖细胞