9个小鼠分成3组后取36个样品做转录组测序可以做多少组合的差异分析
9个小鼠分成3组后取36个样品做转录组测序可以做多少组合的差异分析

转录组测序后差异分析大家应该是都不陌生了,现在的生命科学领域的研究如果不加入一个转录组都会让人很奇怪,慢慢的它的地位开始赶上传统的PCR(聚合酶链反应)或者Western Blot(WB,蛋白质印迹)等技术。
如果一个转录组测序项目只有两个分组,那么简单的单次差异分析即可。但是如果有3分组就麻烦了,简单的理解就是两个处理组去和一个对照组差异,然后这两次差异分析的结果可以去对比一下,比如取交集。如下所示的案例:
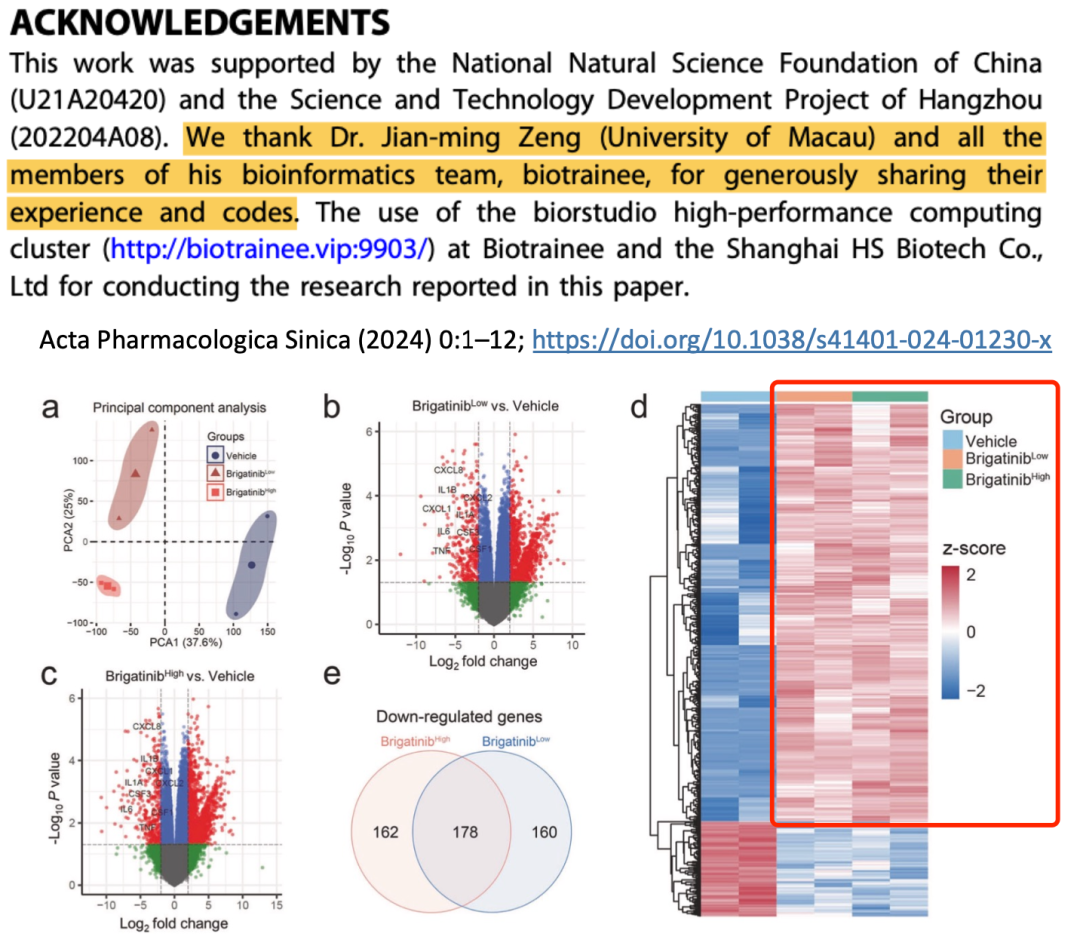
两次差异分析的结果可以取交集
实际上,在进行差异分析时,如果有三组数据(组A、组B和组C),不仅仅是上面的两次差异分析这样的可能性。以下是一些常见的比较组合:
- 组A vs 组B:比较第一组和第二组之间的差异。
- 组A vs 组C:比较第一组和第三组之间的差异。
- 组B vs 组C:比较第二组和第三组之间的差异。
- **组A vs (组B + 组C)**:将组B和组C的数据合并,与组A进行比较。
- **组B vs (组A + 组C)**:将组A和组C的数据合并,与组B进行比较。
- **组C vs (组A + 组B)**:将组A和组B的数据合并,与组C进行比较。
- 每组与所有其他样本的总体比较:在某些情况下,你可能想要比较每组与所有其他样本的总体平均的差异。
- 成对比较:除了上述直接的两两比较,还可以进行成对比较,即比较每一对样本之间的差异。
- 时间序列或剂量反应比较:如果组A、B、C代表时间点或剂量水平,可以比较时间或剂量效应,如组A(时间点1)vs 组B(时间点2)vs 组C(时间点3)。
- 组合比较:可以创建组合的虚拟组,比如将组A和组B的样本合并,然后与组C进行比较,以探究两组间的共同效应或特定效应。
选择哪种比较组合取决于你的研究设计、科学问题和统计测试的要求。在进行差异分析时,重要的是要考虑到多重假设检验的问题,因为多次比较会增加发现假阳性结果的风险。因此,可能需要使用适当的统计校正方法,如Bonferroni校正、Benjamini-Hochberg程序或FDR(False Discovery Rate)控制等。
也就是说,每增加一个分组, 可以选择的分析策略就呈几何的增加,非常恐怖!当然了,大部分情况下,数据分析是需要有生物学背景的指导,我们不可能说是为了分析而分析,比如2019的文章:《Genetic pathway analysis reveals a major role for extracellular matrix organization in inflammatory and neuropathic pain》, 虽然是9个小鼠分成3组后取36个样品做转录组测序,也就是说是12个分组,如下所示:
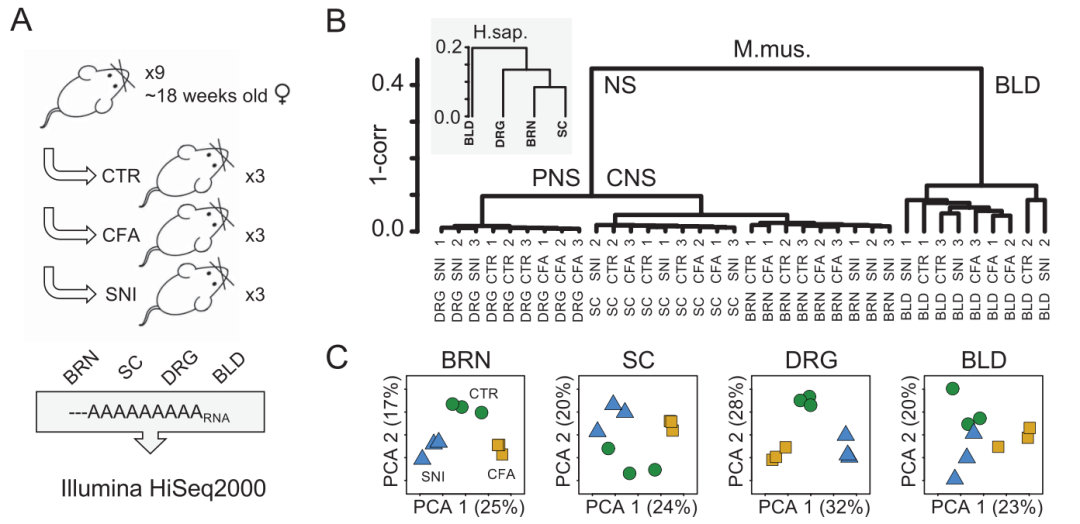
12个分组
首先是小鼠有三分组, 每个组里面是3只小鼠,一个分组是对照的小鼠,另外使用了两种小鼠模型来模拟疼痛状态,:
- 一种是使用完全弗氏佐剂(Complete Freund’s Adjuvant, CFA)诱导的炎症性疼痛(IP)
- 另一种是通过脊髓神经损伤(Spared Nerve Injury, SNI)模型诱导的神经性疼痛(NP)。
然后是涉及到了 小鼠的四个组织(背根神经节、脊髓、大脑和血液):
- whole brain (BRN), spinal cord (SC), dorsal root ganglion (DRG), and whole blood (BLD).
让我们一起看看文章到底是做了多少差异分析,首先是每种组织里面都是可以做疼痛小鼠模型和正常对照小鼠的差异:
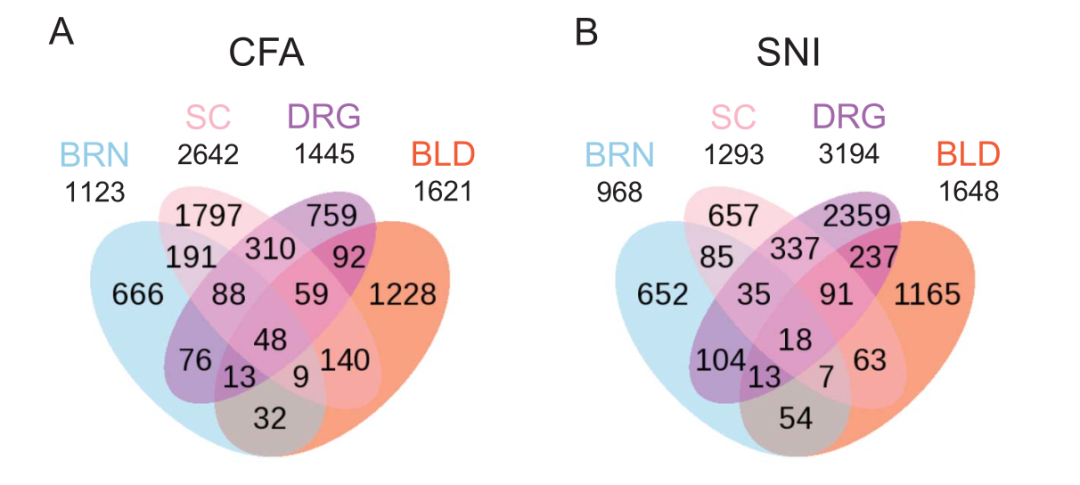
做疼痛小鼠模型和正常对照小鼠的差异
上面的两种疼痛小鼠模型还可以在每个组织里面独立的取交集:
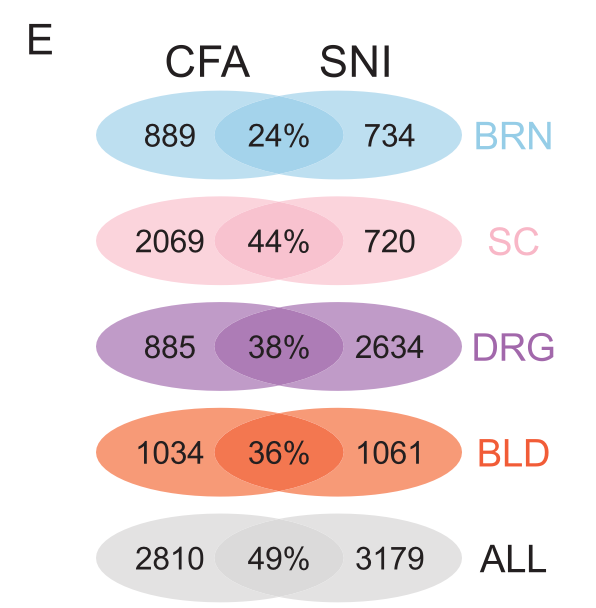
每个组织里面独立的取交集
其实这个时候或许mfuzz或者wgcna这样的针对基因进行直接分组的算法可能会好一点,这个文章就没有做,未必就不能作为一个公共数据挖掘的课题啦, 当然了,如果是结合类似的实验设计的单细胞转录组数据就更容易得出有意义的生物学结论了,这一切的前提是有生物学背景而不仅仅是跑生物信息学软件工具啦。
而且,这样的公共数据集理论上是足够多的,因为疼痛研究领域也不小众,比如上面的文章就对比了3个公共数据集:
- rat spinal cord 7 days SNI vs sham (GEO set GSE18803),
- rats DRG ipsilateral 7 days SNI vs sham (GEO set GSE15041),
- rats DRG 3 days CFA vs sham (GEO set GSE38859).