单细胞系列教程:质控实战(五)
原创
1. 学习目标
- 构建质量控制指标并评估数据质量
- 适当的应用过滤器去除低质量的细胞
2. 过滤目标
- 过滤数据以仅包含高质量的真实细胞,以便在对细胞进行聚类时更容易识别不同的细胞类型
- 对一些不合格样品的数据进行检查,试图查询其不合格的原因
3. 挑战
- 从少量复杂的细胞中描绘出质量较差的细胞
- 选择合适的过滤阈值,以便在不去除生物学相关细胞类型的情况下保留高质量的细胞
4. 质量标准
当数据加载到 Seurat 并创建初始对象时,会为计数矩阵中的每个单元组装一些基本元数据。要仔细查看此元数据,查看存储在 merge_seurat对象的 meta.data中的数据帧:
# 查看元数据
View(merged_seurat@meta.data) # 具体介绍见质控准备章节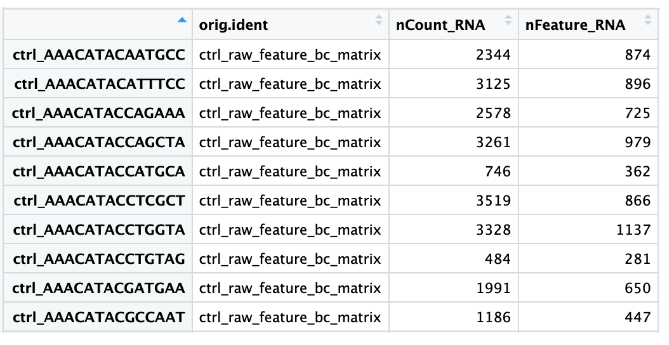
为了可视化质量控制分析情况,需要计算一些额外的指标。这些包括:
number of genes detected per UMI:这个指标让了解数据集的复杂性(每个UMI检测到越多基因,数据越复杂)mitochondrial ratio:该指标将提供来自线粒体基因的细胞读数百分比
5. Novelty score
这个值很容易计算,取每个细胞检测到的基因数量的log10 和每个细胞的 UMI数量的log10,然后将 log10的基因数量除以UMI的log10数量。
# 将每个单元格的每个 UMI 的基因数添加到元数据
merged_seurat$log10GenesPerUMI <- log10(merged_seurat$nFeature_RNA) / log10(merged_seurat$nCount_RNA)6. 线粒体率
Seurat 有一个方便的功能,可以计算映射到线粒体基因的转录本比例。PercentageFeatureSet()函数接受一个模式参数,并在数据集中的所有基因标识符中搜索该模式。由于正在寻找线粒体基因,因此搜以“MT-”模式开头的任何基因标识符。对于每个细胞,该函数获取属于“Mt-”集的所有基因(特征)的计数总和,然后除以所有基因(特征)的计数总和。该值乘以 100 以获得百分比值。
# 计算百分比
merged_seurat$mitoRatio <- PercentageFeatureSet(object = merged_seurat, pattern = "^MT-") # 该模式 ^MT- 应该根据自己的数据集进行修改
merged_seurat$mitoRatio <- merged_seurat@meta.data$mitoRatio / 100现在已经具备了评估数据所需的质量指标。但是,希望在元数据中包含一些有用的附加信息,包括单元 ID 和条件信息。首先通过从Seurat对象中提取 meta.data 来创建元数据:
# 创建元数据
metadata <- merged_seurat@meta.data
# 将 Cell ID 添加到元数据
metadata$cells <- rownames(metadata)
# 创建样本列
metadata$sample <- NA
metadata$sample[which(str_detect(metadata$cells, "^ctrl_"))] <- "ctrl"
metadata$sample[which(str_detect(metadata$cells, "^stim_"))] <- "stim"
# 重命名列
metadata <- metadata %>% dplyr::rename(seq_folder = orig.ident,
nUMI = nCount_RNA,
nGene = nFeature_RNA)
# 最终结果如下图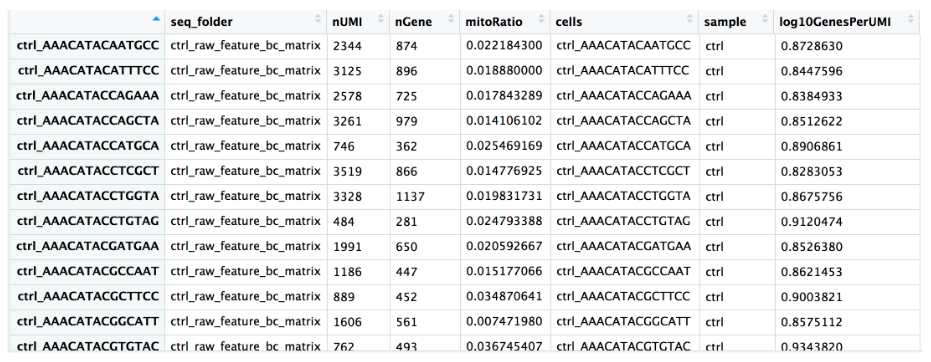
# 将更新的元数据保存到` Seurat` 对象
merged_seurat@meta.data <- metadata
# 保存为`.RData`
save(merged_seurat, file="data/merged_filtered_seurat.RData")
# 结果如下
7. 质量评估指标
下面将评估以下各种指标,然后决定哪些cells质量低,应从分析中删除
Cell counts
细胞计数由检测到的独特细胞条形码的数量决定。对于本实验,预计在 12,000 -13,000 个细胞之间。
在理想情况下,会期望唯一细胞条形码的数量与加载的细胞数量相对应。然而,情况并非如此,因为细胞的捕获率只是加载的一部分。例如,与 50-60% 之间的 10X 相比,inDrops 细胞捕获效率更高(70-80%)。
细胞数量也可能因protocol而异,产生的细胞数量远高于加载的数量。例如,在inDrops protocol期间,细胞条形码存在于水凝胶中,这些水凝胶与单个细胞和裂解/反应混合物一起封装在液滴中。虽然每个水凝胶都应该有一个与之相关的细胞条形码,但有时水凝胶可以有多个细胞条形码。同样,使用10X protocol时,有可能仅在乳液液滴 (GEM) 中获得带条形码的珠子,而没有实际的细胞。除了死亡细胞的存在之外,这两者都可能导致比细胞更多的细胞条形码。
# 可视化每个样本的细胞计数
metadata %>%
ggplot(aes(x=sample, fill=sample)) +
geom_bar() +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) +
theme(plot.title = element_text(hjust=0.5, face="bold")) +
ggtitle("NCells")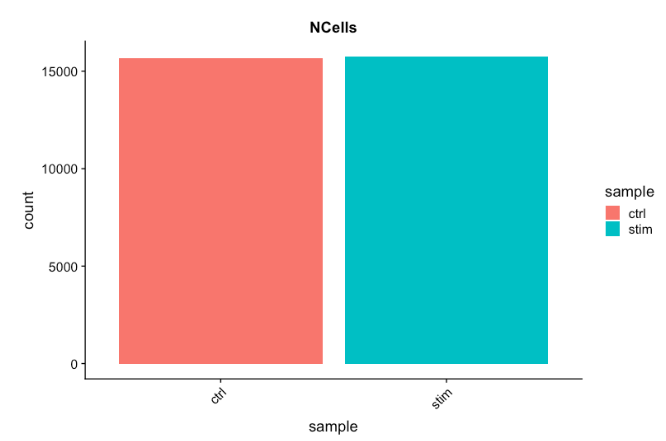
每个样本超过 15,000 个细胞,这比预期的 12-13,000 个要多。很明显,可能存在一些垃圾“细胞”。
UMI counts per cell
每个spot的 UMI 计数通常应高于 500,这是预期的。如果UMI计数在 500-1000 计数之间,是可用的,但可能应该对细胞进行更深入的测序。
# 可视化 UMIs/transcripts per cell 数量
metadata %>%
ggplot(aes(color=sample, x=nUMI, fill= sample)) +
geom_density(alpha = 0.2) +
scale_x_log10() +
theme_classic() +
ylab("Cell density") +
geom_vline(xintercept = 500)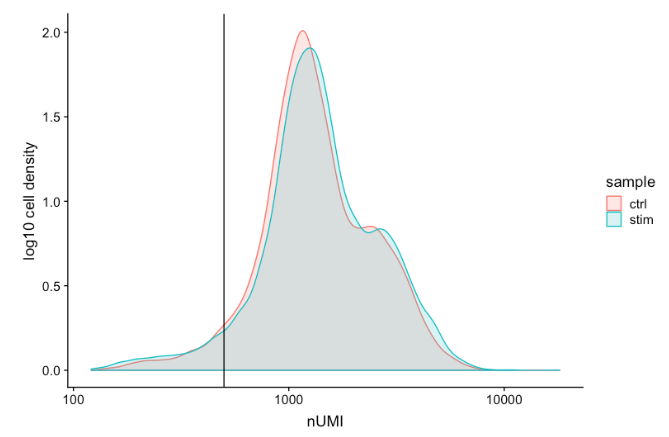
可以看到,两个样本中的大多数细胞都有 1000 或更高的 UMI。
Genes detected per cell
对基因检测的期望与对UMI 检测相似,尽管它可能比 UMI低一点。对于高质量数据,比例直方图应包含一个代表被封装细胞的大峰。如果看到主峰左侧有一个小肩,或者细胞的双峰分布,这可能表明有一些问题。可能有一组单元由于某种原因失败了。也可能是存在生物学上不同类型的细胞。
# 通过直方图可视化每个细胞检测到的基因分布
metadata %>%
ggplot(aes(color=sample, x=nGene, fill= sample)) +
geom_density(alpha = 0.2) +
theme_classic() +
scale_x_log10() +
geom_vline(xintercept = 300)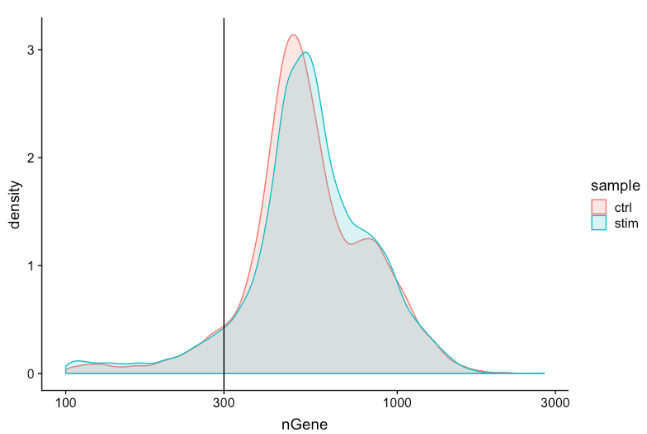
Complexity(novelty score)
可以使用一种名为novelty score的来评估每个细胞 RNA 种类的复杂程度。novelty score是通过 nGenes 与nUMI的比率来计算的。如果有很多捕获的转录本(高nUMI)并且在一个细胞中检测到少量基因,这可能意味着只捕获了少量基因,并且只是一遍又一遍地从这些较少数量的基因中测序转录本。这些低复杂性(低novelty score)的细胞可能代表特定的细胞类型(即缺乏典型转录组的红细胞),或者可能是由于人为因素或污染造成的。一般来说,预计优质细胞的novelty score高于 0.80。
# 通过可视化每个 UMI 检测到的基因来可视化基因表达的整体复杂性(novelty score分)
metadata %>%
ggplot(aes(x=log10GenesPerUMI, color = sample, fill=sample)) +
geom_density(alpha = 0.2) +
theme_classic() +
geom_vline(xintercept = 0.8)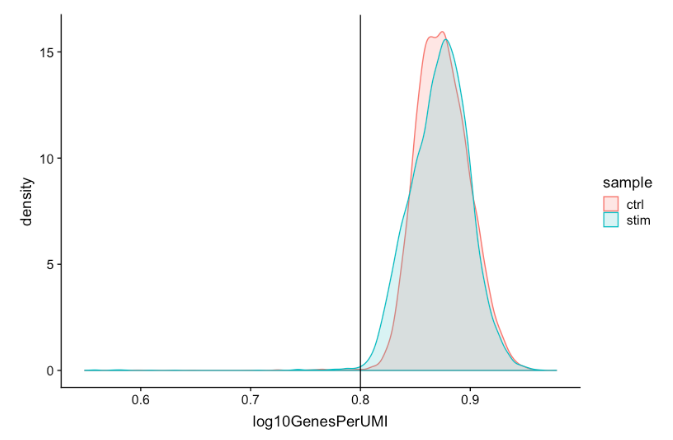
Mitochondrial counts ratio
该指标可以确定是否存在大量来自死亡或垂死细胞的线粒体污染。将线粒体计数的劣质样本定义为超过 0.2 线粒体比率标记的细胞。
# 可视化每个细胞检测到的线粒体基因表达分布
metadata %>%
ggplot(aes(color=sample, x=mitoRatio, fill=sample)) +
geom_density(alpha = 0.2) +
scale_x_log10() +
theme_classic() +
geom_vline(xintercept = 0.2)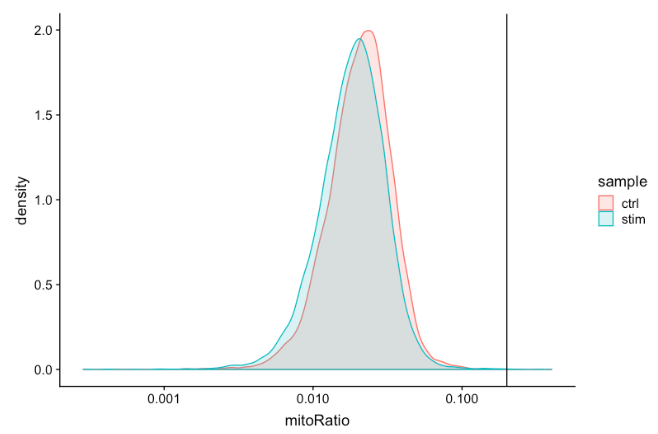
Joint filtering effects
孤立地考虑这些QC 指标中的任何一个都可能导致对信号的误解。例如,线粒体计数比例较高的细胞可能参与呼吸过程,并且可能是想要保留的细胞。同样,其他指标可以有其他生物学解释。执行QC时的一般经验法则是将单个指标的阈值设置为尽可能宽松,并始终考虑这些指标的联合影响。通过这种方式,可以降低过滤掉任何活细胞群的风险。
经常一起评估的两个指标是UMI 的数量和每个细胞检测到的基因数量。在这里,绘制了基因数量与线粒体读数分数着色的 UMI数量的关系。联合可视化计数和基因阈值并额外覆盖线粒体分数,得出每个细胞质量的总结图。
# 可视化检测到的基因与 UMI 数量之间的相关性,并确定是否存在大量基因/UMI 数量少的细胞
metadata %>%
ggplot(aes(x=nUMI, y=nGene, color=mitoRatio)) +
geom_point() +
scale_colour_gradient(low = "gray90", high = "black") +
stat_smooth(method=lm) +
scale_x_log10() +
scale_y_log10() +
theme_classic() +
geom_vline(xintercept = 500) +
geom_hline(yintercept = 250) +
facet_wrap(~sample)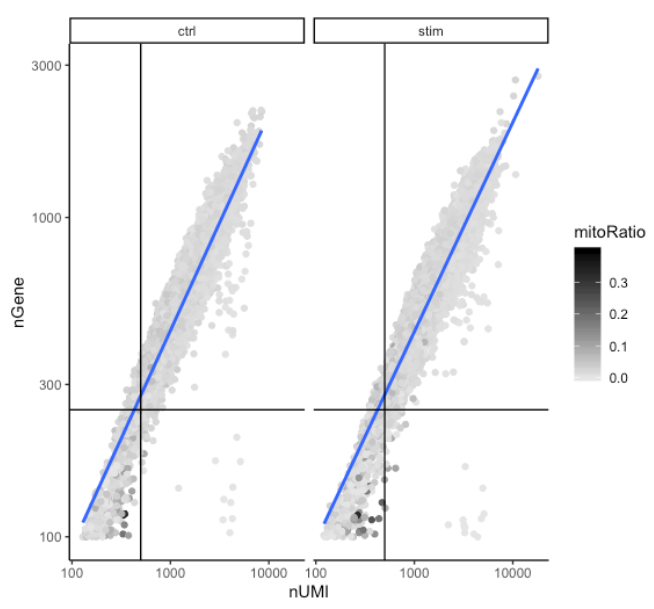
好的细胞通常会表现出每个细胞更多的基因和更多的UMI(图的右上象限)。质量差的细胞可能每个细胞的基因和 UMI 较低,并且对应于图左下象限中的数据点。通过该图,评估了线的斜率,以及该图右下象限中数据点的任何散布。这些细胞具有大量的UMI,但只有少数基因。这些可能是垂死的细胞,但也可能代表低复杂性细胞类型(即红细胞)的群体。
线粒体分数仅在很少(颜色较深的数据点)的特别低计数的细胞中较高。这可能表明其细胞质 mRNA 已通过破裂的膜泄漏出来的受损/垂死细胞,因此,只有位于线粒体中的 mRNA 仍然是保守的。可以从图中看到,这些细胞被计数和基因数阈值过滤掉了。
8. 过滤
Cell-level过滤
现在已经可视化了各种指标,可以决定要使用的阈值,这将导致删除低质量的单元格。前面提到的建议通常是一个粗略的指导,具体的实验需要告知选择的确切阈值。下面将使用以下阈值:
- nUMI > 500
- nGene > 250
- log10GenesPerUMI > 0.8
- mitoRatio < 0.2
为了过滤,将回到 Seurat对象并使用subset()函数:
# 使用选定的阈值过滤掉低质量的细胞
filtered_seurat <- subset(x = merged_seurat,
subset= (nUMI >= 500) &
(nGene >= 250) &
(log10GenesPerUMI > 0.80) &
(mitoRatio < 0.20))Gene-level过滤
在数据中,将有许多计数为零的基因。这些基因可以显着降低细胞的平均表达,因此将从数据中删除它们。首先确定每个细胞中哪些基因的计数为零:
# 提取计数
counts <- GetAssayData(object = filtered_seurat, slot = "counts")
# 输出一个逻辑矩阵,为每个基因指定每个细胞的计数是否超过零
nonzero <- counts > 0现在,将按novelty score进行一些过滤。如果一个基因只在少数几个细胞中表达,那么它并不是特别有意义,因为它仍然会降低所有其他不表达它的细胞的平均值。选择只保留在 10 个或更多细胞中表达的基因细胞。通过使用此过滤器,将有效去除所有细胞中计数为零的基因。
# 对所有 TRUE 值求和,如果每个基因超过 10 个 TRUE 值,则返回 TRUE
keep_genes <- Matrix::rowSums(nonzero) >= 10
# 只保留那些在超过 10 个细胞中表达的基因
filtered_counts <- counts[keep_genes, ]最后,获取这些过滤计数并创建一个新的 Seurat 对象以进行下游分析。
# 重新分配给过滤后的 Seurat 对象
filtered_seurat <- CreateSeuratObject(filtered_counts, meta.data = filtered_seurat@meta.data)9. 重新评估
执行过滤后,建议回顾指控指标以确保数据符合预期并且有利于下游分析。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。