Seurat软件学习10-SCTransformV2版本的运行方式
原创Seurat软件学习10-SCTransformV2版本的运行方式
原创
Seurat软件学习1-多个模型得数据进行整合:/developer/article/2130078
Seurat软件学习2-scrna数据整合分析:/developer/article/2131431
Seurat软件学习3-scrna数据整合分析注释数据集:/developer/article/2133583
Seurat软件学习4-使用RPCA进行快速整合数据集:/developer/article/2134684
Seurat软件学习5-scRNA-Seq和scATAC-Seq数据整合:/developer/article/2136814
Seurat软件学习6-多模型参考映射的方法:/developer/article/2144475
Seurat软件学习7-同胞多组学结合方法-WNN:/developer/article/2152008
Seurat软件学习8-不同细胞类型样本的分析流程:/developer/article/2191271
Seurat软件学习9-sctransform的使用:/developer/article/2208107?areaSource=&traceId=
TL;DR
前期经过sctransform,可以对scRNA-seq数据集进行更好的均一化处理。同时目前根据对59个scRNA-seq数据集的广泛分析,对相关的V1的版本进行更新,V2版本进行了一系列的技术更新,包括更新提高了速度和内存消耗、参数估计的稳定性、可变特征的识别,以及进行下游差异表达分析的能力。
用户可以从Github上安装sctransform v2,并通过vst.flavor参数调用使用更新的软件方法。
# install Seurat from Github (automatically updates sctransform)
devtools::install_github("satijalab/seurat", ref = "develop")
# invoke sctransform
object <- SCTransform(object, vst.flavor = "v2")Introduction
单细胞RNA-seq与其他数据相比,差异来源主要是包括细胞状态的生物变异及试验处理之间引入的技术从差异。因此通过使用广义线性模型或基于似然的方法进行预处理和下游分析时可以更大的解决上述的问题。
在本节中,通过使用sctransform v2的流程脚本,对处于静止或干扰素刺激状态的人类免疫细胞(PBMC)进行比较分析。在本节中,使用sctransform-v2的归一化来进行下面的分析。
1.建立一个 "整合 "的数据分析
2.用于下游分析比较数据集,找到细胞类型对刺激的特定反应
3.获得在对照和刺激的细胞中保守的marker gene
Install sctransform
首先在Github上安装sctransform v2。继而安装glmGamPoi包,它可以大大改善分析速度。
# install glmGamPoi
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("glmGamPoi")
# install sctransform from Github
devtools::install_github("satijalab/sctransform", ref = "develop")Setup the Seurat objects
library(Seurat)
library(SeuratData)
library(patchwork)
library(dplyr)
library(ggplot2)该数据集可通过SeuratData软件包获得相关的数据。
# install dataset
InstallData("ifnb")
# load dataset
LoadData("ifnb")
# split the dataset into a list of two seurat objects (stim and CTRL)
ifnb.list <- SplitObject(ifnb, split.by = "stim")
ctrl <- ifnb.list[["CTRL"]]
stim <- ifnb.list[["STIM"]]Perform normalization and dimensionality reduction
为了进行均一化,在选用SCTransform进行分析时主要用vst.flavor="v2 "来调用v2进行相关分析,这与最初在Hafemeister和Satija, 2019中介绍的方法进行了改进。
1.主要是将GLM的斜率参数固定为ln(10),并按照Lause等人的建议将log10(总UMI)作为预测因子。
2.利用改进的参数估计程序,减少了对表达量很低的基因进行GLM模型拟合时产生的不确定性和偏差。
3.在计算Pearson残差时,对基因水平的标准偏差设置了一个下限。这可以降低表达量极低的基因(只有1-2个检测到的UMI)出现高的皮尔森残差。
# normalize and run dimensionality reduction on control dataset
ctrl <- SCTransform(ctrl, vst.flavor = "v2", verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE) %>%
RunUMAP(reduction = "pca", dims = 1:30, verbose = FALSE) %>%
FindNeighbors(reduction = "pca", dims = 1:30, verbose = FALSE) %>%
FindClusters(resolution = 0.7, verbose = FALSE)
p1 <- DimPlot(ctrl, label = T, repel = T) + ggtitle("Unsupervised clustering")
p2 <- DimPlot(ctrl, label = T, repel = T, group.by = "seurat_annotations") + ggtitle("Annotated celltypes")
p1 | p2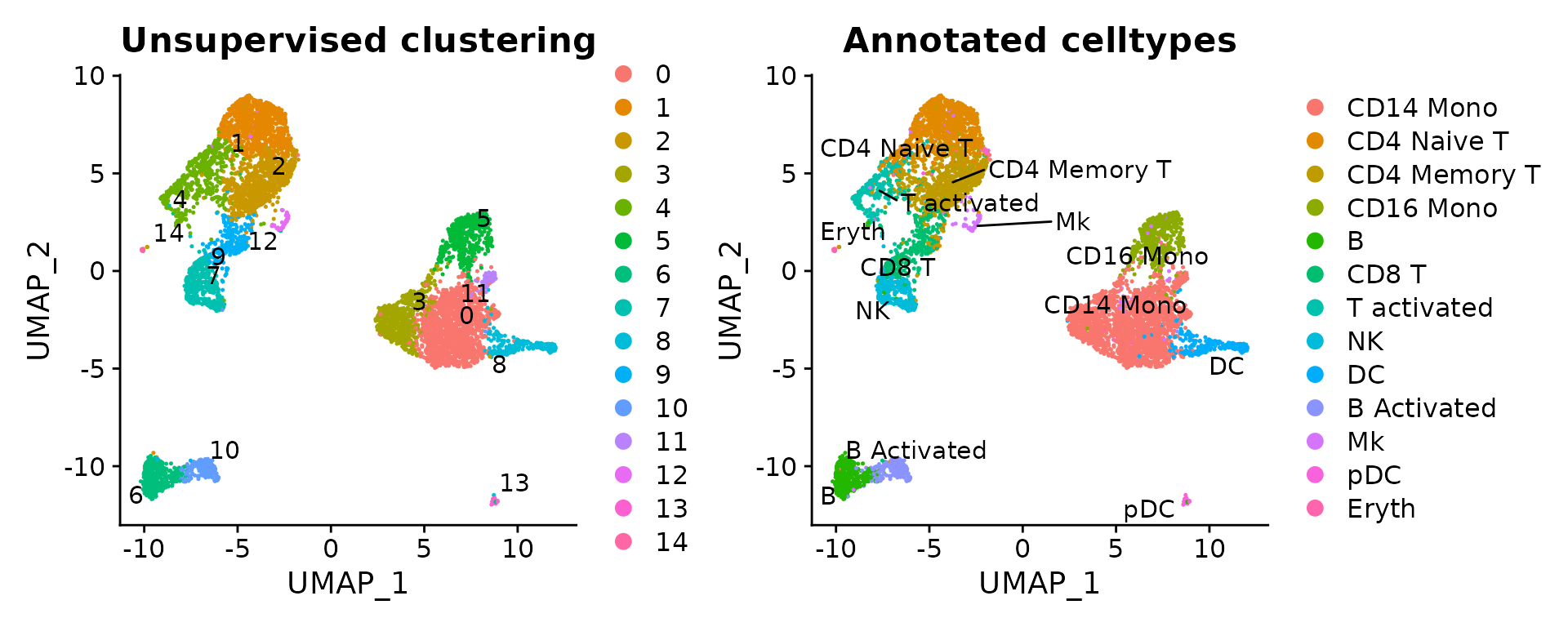
Perform integration using pearson residuals
在使用SelectIntegrationFeatures()选择信息特征列表后,使用PrepSCTIntegration()函数进行整合。
stim <- SCTransform(stim, vst.flavor = "v2", verbose = FALSE) %>%
RunPCA(npcs = 30, verbose = FALSE)
ifnb.list <- list(ctrl = ctrl, stim = stim)
features <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 3000)
ifnb.list <- PrepSCTIntegration(object.list = ifnb.list, anchor.features = features)使用FindIntegrationAnchors()函数进行两个数据集的整合,该函数将Seurat对象的列表作为输入,并使用这些锚点将两个数据集进行整合。
##这里出现了sct的normalization的要求,如果没有,就是默认的normalization方法,前面的解析中也有
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, normalization.method = "SCT",
anchor.features = features)
immune.combined.sct <- IntegrateData(anchorset = immune.anchors, normalization.method = "SCT")Perform an integrated analysis
现在可以对所有细胞进行一次整合分析。
immune.combined.sct <- RunPCA(immune.combined.sct, verbose = FALSE)
immune.combined.sct <- RunUMAP(immune.combined.sct, reduction = "pca", dims = 1:30, verbose = FALSE)
immune.combined.sct <- FindNeighbors(immune.combined.sct, reduction = "pca", dims = 1:30)
immune.combined.sct <- FindClusters(immune.combined.sct, resolution = 0.3)
###对相关的结果进行可视化
DimPlot(immune.combined.sct, reduction = "umap", split.by = "stim")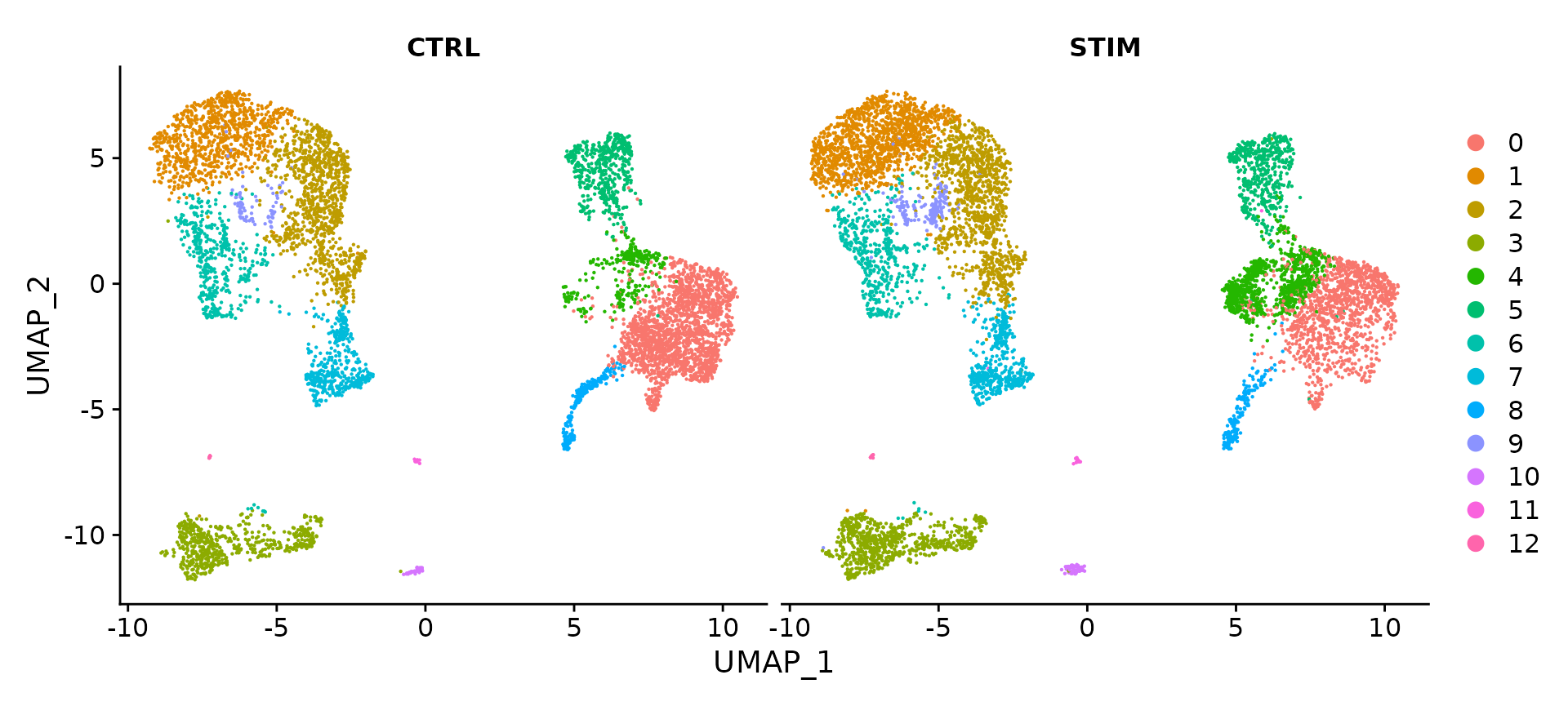
选择不同的group.by 进行可视化不同条件下的umap图。
p1 <- DimPlot(immune.combined.sct, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined.sct, reduction = "umap", group.by = "seurat_clusters", label = TRUE,
repel = TRUE)
p3 <- DimPlot(immune.combined.sct, reduction = "umap", group.by = "seurat_annotations", label = TRUE,
repel = TRUE)
p1 | p2 | p3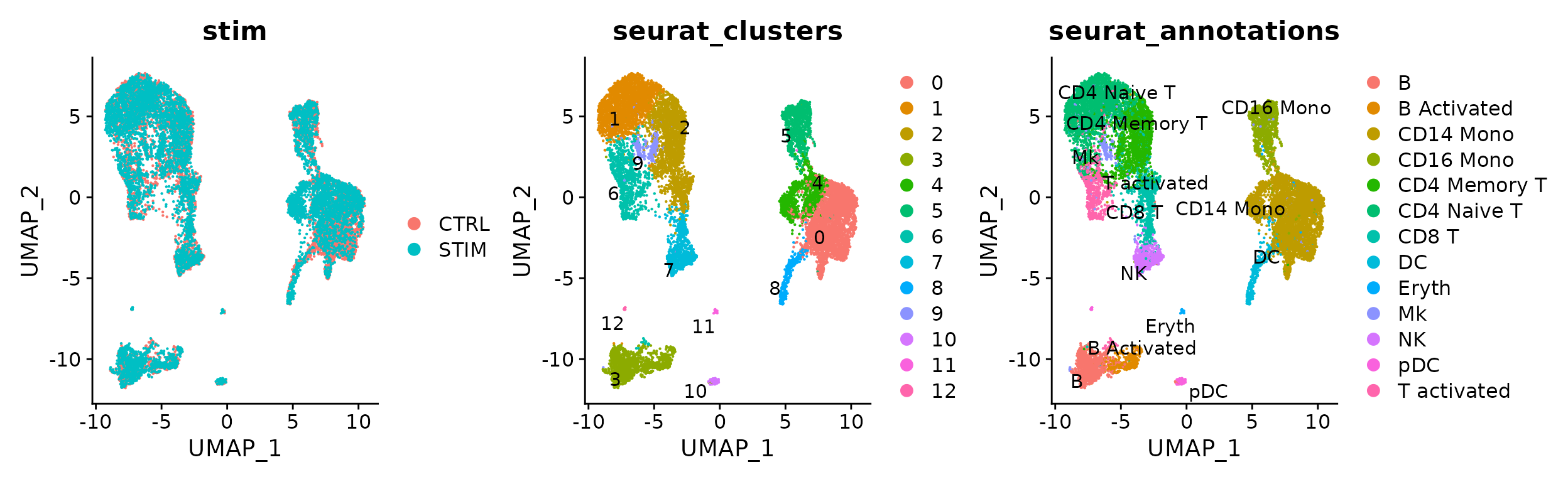
Identify differential expressed genes across conditions
为了查看相同的细胞在不同条件下的变化,在前面通过对meta.data的选择,来对后面的结果进行相关的可视化。
immune.combined.sct$celltype.stim <- paste(immune.combined.sct$seurat_annotations, immune.combined.sct$stim,
sep = "_")
Idents(immune.combined.sct) <- "celltype.stim"为了运行差异表达,利用了储存在SCT矩阵的 "校正计数"。通过将所有细胞的测序深度设置为一个固定值并反转所学的正则化负二项回归模型来获得校正计数。在进行差异表达之前,首先运行PREPSCTFindMarkers,以确保counts值被正确设置。然后使用FindMarkers(assay="SCT")来寻找差异表达的基因。这里分析的主要目的是确定受刺激的B细胞和对照B细胞之间差异表达基因。
immune.combined.sct <- PrepSCTFindMarkers(immune.combined.sct)
b.interferon.response <- FindMarkers(immune.combined.sct, assay = "SCT", ident.1 = "B_STIM", ident.2 = "B_CTRL",
verbose = FALSE)
head(b.interferon.response, n = 15)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## ISG15 1.505693e-159 3.5088882 0.998 0.229 2.003475e-155
## IFIT3 4.128835e-154 2.5684404 0.961 0.052 5.493827e-150
## IFI6 2.479476e-153 2.4602058 0.965 0.076 3.299190e-149
## ISG20 9.385626e-152 2.5618077 1.000 0.666 1.248851e-147
## IFIT1 2.447118e-139 2.1328142 0.904 0.029 3.256136e-135
## MX1 2.111944e-124 1.9311875 0.900 0.115 2.810153e-120
## LY6E 2.930414e-122 1.8496310 0.898 0.150 3.899209e-118
## TNFSF10 1.104024e-112 1.7817968 0.785 0.020 1.469014e-108
## IFIT2 3.491368e-108 1.7594656 0.783 0.037 4.645615e-104
## B2M 3.405403e-98 0.5980104 1.000 1.000 4.531229e-94
## IRF7 1.114291e-96 1.4721297 0.834 0.187 1.482675e-92
## PLSCR1 3.364901e-96 1.4495896 0.783 0.111 4.477338e-92
## UBE2L6 1.155610e-85 1.3221839 0.849 0.295 1.537655e-81
## CXCL10 5.689834e-84 3.0310329 0.639 0.010 7.570893e-80
## PSMB9 2.304426e-81 1.2355834 0.937 0.568 3.066269e-77如果在运行PrepSCTFindMarkers()之后在原始对象的子集上运行,FindMarkers()应该在recorrect_umi = FALSE的情况下调用,使用现在规定的counts值。
immune.combined.sct.subset <- subset(immune.combined.sct, idents = c("B_STIM", "B_CTRL"))
b.interferon.response.subset <- FindMarkers(immune.combined.sct.subset, assay = "SCT", ident.1 = "B_STIM",
ident.2 = "B_CTRL", verbose = FALSE, recorrect_umi = FALSE)
##对相关的结果进行可视化
Idents(immune.combined.sct) <- "seurat_annotations"
DefaultAssay(immune.combined.sct) <- "SCT"
FeaturePlot(immune.combined.sct, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3,
cols = c("grey", "red"))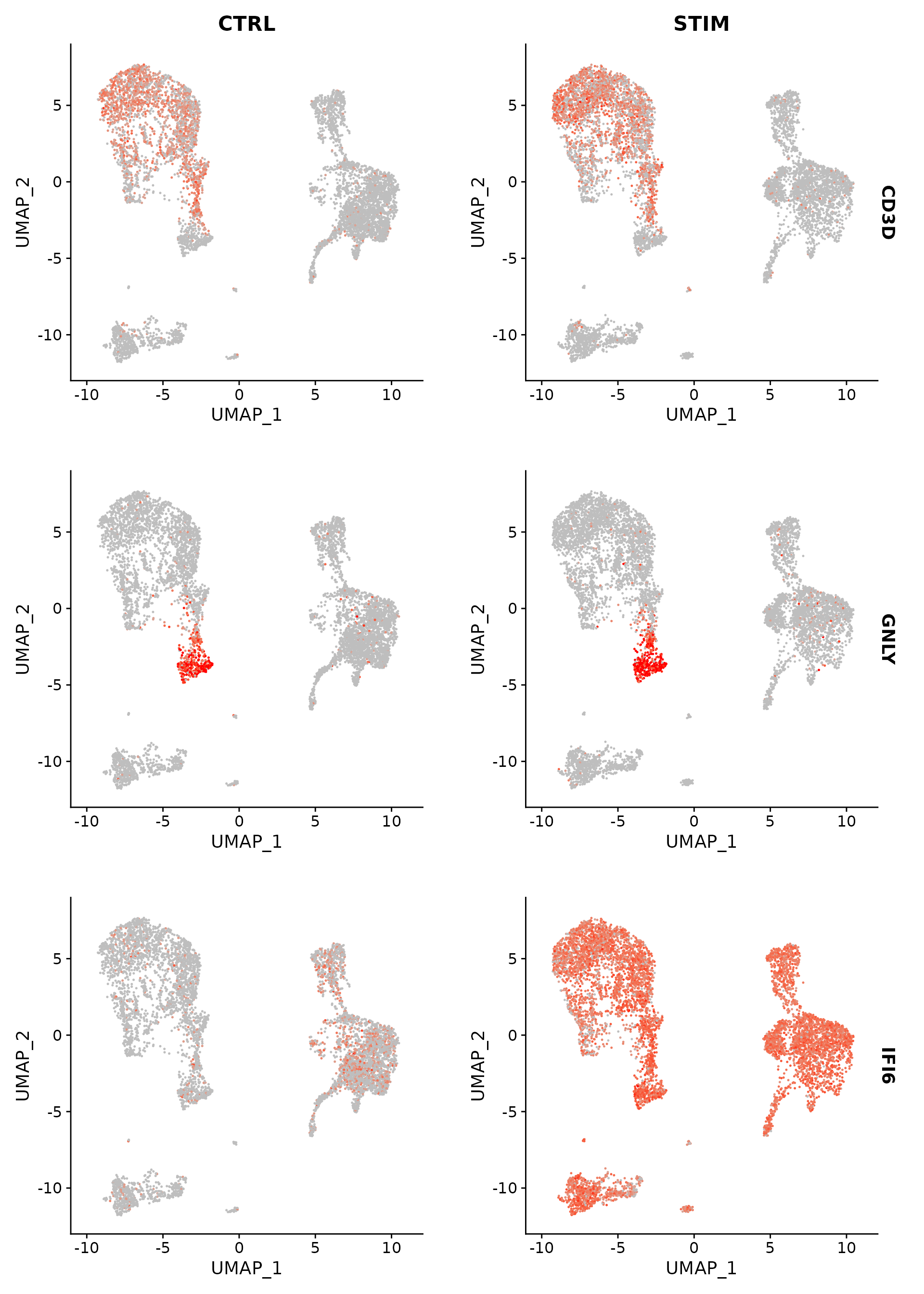
plots <- VlnPlot(immune.combined.sct, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim",
group.by = "seurat_annotations", pt.size = 0, combine = FALSE)
wrap_plots(plots = plots, ncol = 1)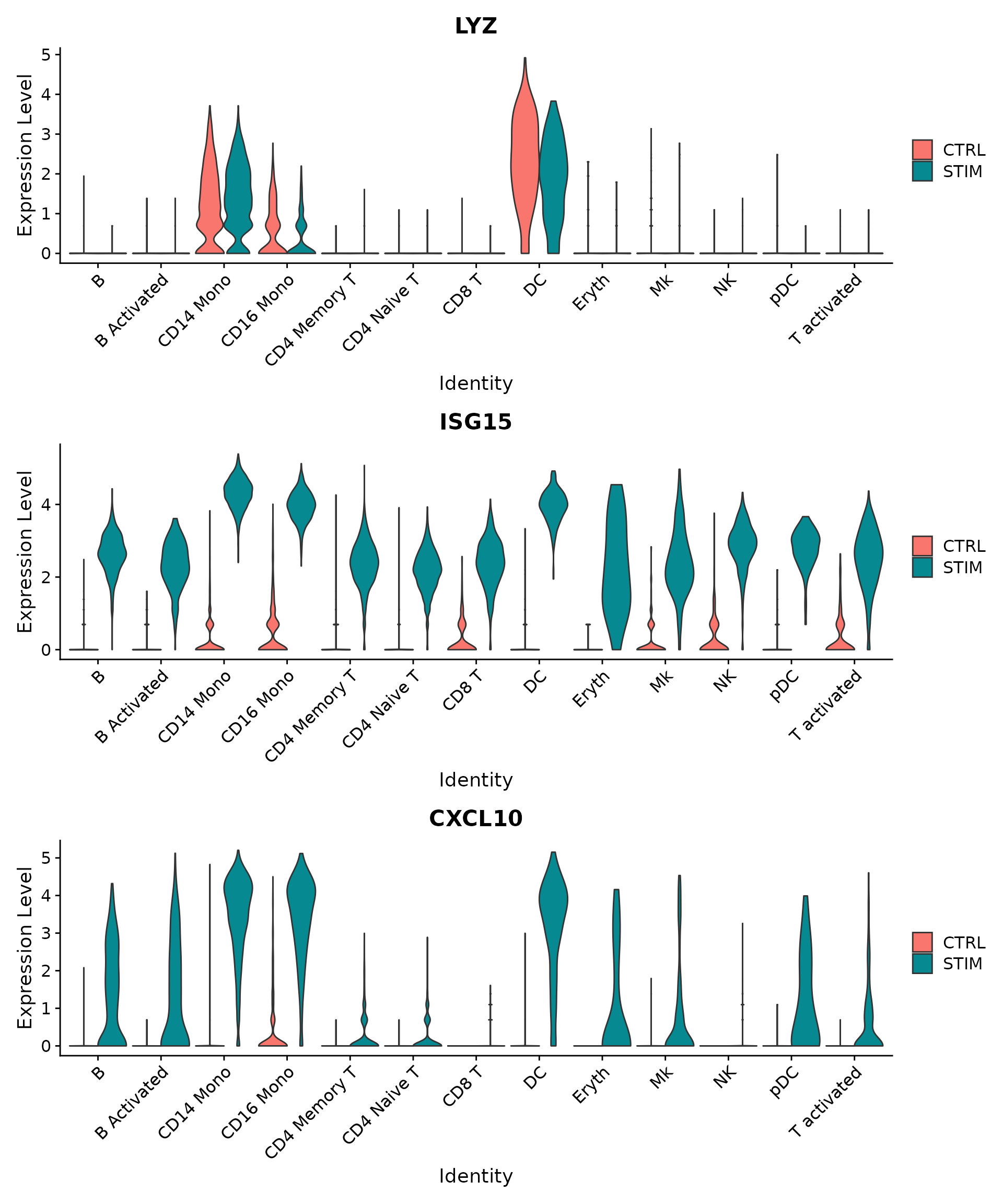
Identify conserved cell type markers
为了识别不同条件下保守的典型细胞类型标记基因,选用FindConservedMarkers()函数。这个函数对每个数据集/组进行差异基因表达测试,并使用MetaDE R软件包中的元分析方法对P值进行组合。例如,我们可以确定在NK细胞中,无论任何条件下都是保守的标志物的基因。注意,PrepSCTFindMarkers命令不需要再次运行。
nk.markers <- FindConservedMarkers(immune.combined.sct, assay = "SCT", ident.1 = "NK", grouping.var = "stim",
verbose = FALSE)
head(nk.markers)
## CTRL_p_val CTRL_avg_log2FC CTRL_pct.1 CTRL_pct.2 CTRL_p_val_adj
## GNLY 0 4.2126974 0.943 0.045 0
## FGFBP2 0 1.2675614 0.500 0.020 0
## CLIC3 0 1.3891367 0.601 0.024 0
## CTSW 0 1.0842918 0.537 0.029 0
## KLRD1 0 0.9382079 0.507 0.019 0
## KLRC1 0 0.7310252 0.379 0.004 0
## STIM_p_val STIM_avg_log2FC STIM_pct.1 STIM_pct.2 STIM_p_val_adj
## GNLY 0.000000e+00 4.3429777 0.956 0.056 0.000000e+00
## FGFBP2 3.703265e-160 0.6174405 0.259 0.015 4.927564e-156
## CLIC3 0.000000e+00 1.5325425 0.623 0.030 0.000000e+00
## CTSW 0.000000e+00 1.2323017 0.592 0.035 0.000000e+00
## KLRD1 0.000000e+00 1.0502928 0.555 0.026 0.000000e+00
## KLRC1 0.000000e+00 0.7401034 0.374 0.005 0.000000e+00
## max_pval minimump_p_val
## GNLY 0.000000e+00 0
## FGFBP2 3.703265e-160 0
## CLIC3 0.000000e+00 0
## CTSW 0.000000e+00 0
## KLRD1 0.000000e+00 0
## KLRC1 0.000000e+00 0原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。