生信干货~SRA下载后批量处理Counts文件
生信干货~SRA下载后批量处理Counts文件

新起点 国自然终于都交完了~开始更新生信干货教程~~~
在这之前先看下面的教程
总结 从零到壹:10元转录组分析小结~干货~ 然后,重点看批量处理数据的技巧~从零到壹:10元转录组分析 从零到壹:10元转录组分析~硬盘不够用咋办 从零到壹:10元~Mapping神器STAR的安装及用 从零到壹:从SRA下载到分析~纯干货 10元转录组分析:这次真的是干货了~灰常干
得到ReadsPerGene数据后
得到每个基因的Counts数之后,你需要将这些不同文件中的提取出来,以制备DEseq2所需要的原始文件,组数少的情况下很好吧,看好第几列、第几行,用R语言按照下面的命令就可以x<-Counts[-(1:4),2] #去掉的1到4行,选取第2列然后用cbind把所有x并在一起就OK了。
但是数量巨大怎么办
比如以下这样的300+样本
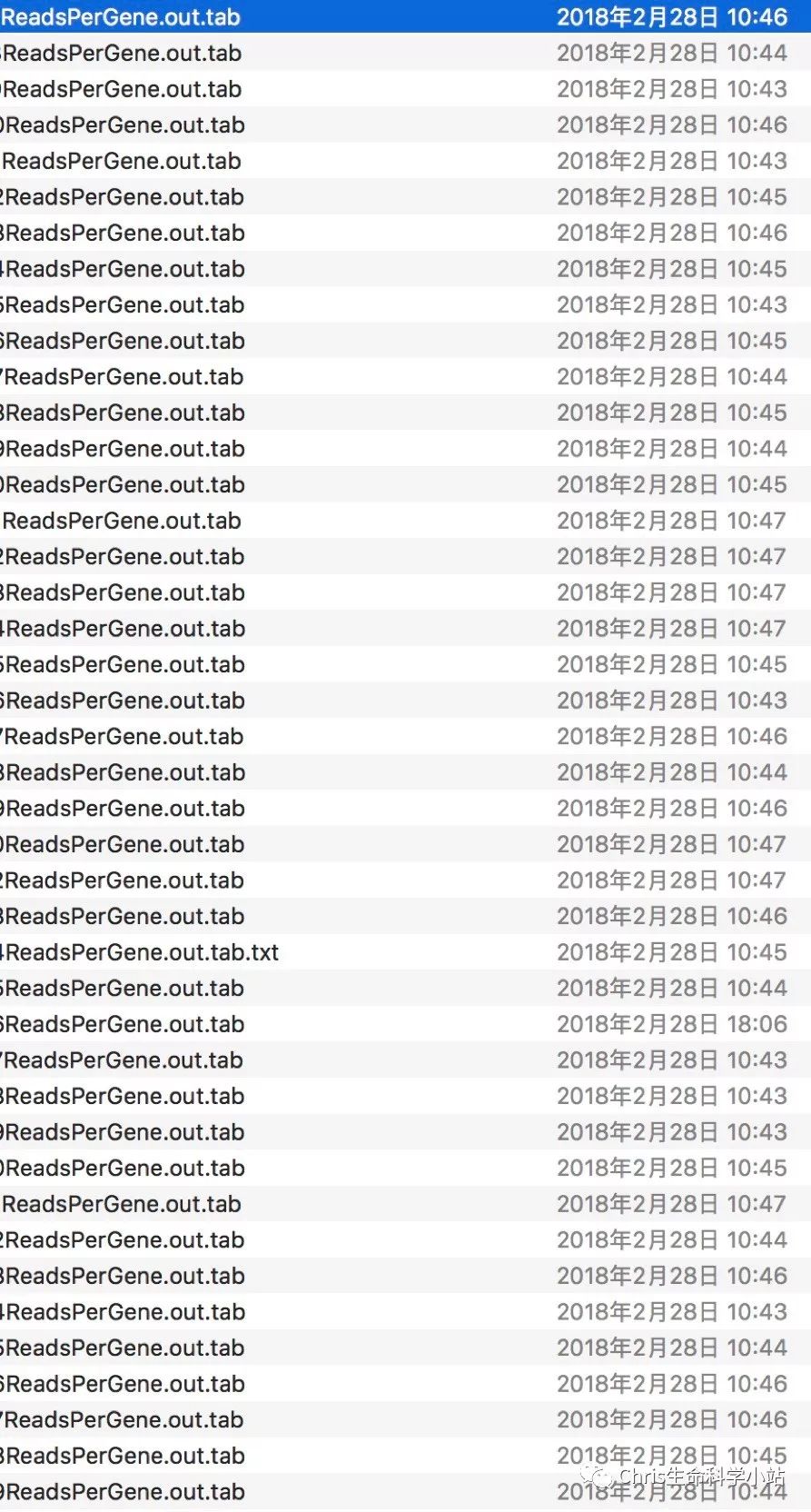
"少废话,来干货~"
将R语言工作环境设置为这些文件所在文件夹
注意这些文件夹中不能有其他文件
如果你的样本是链特异性(Reverse)测序
“啥是链特异性,需要解释的留言”
用下面这组命令
library(stringr)
library(dplyr)
fall <- dir()
data.out<-df.use[1:4,]
for (fnow in fall) {
str <- str_sub(fnow, start = 4, end = 10)
num <- as.numeric(str)
df.read <- read.table(fnow)
df.use <- data.frame(v1 = df.read$V1,v4=df.read$V4)
str_c<-str_c('SRR',str)
colnames(df.use)<-c('V1',str_c)
data.out <- full_join(data.out, df.use,by="V1")
}
data.out1<-data.out[-(1:4),-2] #这个是对data.out修整
write.csv(data.out1, file = 'F:/out.csv')
data.out1 就是DEseq2包中需要用的文件
之后的就分析吧
~~~~~~~
未完待续