生物信息学常见数据格式以及文本处理(grep/sed/awk)
原创生物信息学常见数据格式以及文本处理(grep/sed/awk)
原创
一、生物信息学常见数据格式(一)fasta 格式(*.fa)
可以表示多肽序列或者核酸序列,包括id行和序列行。

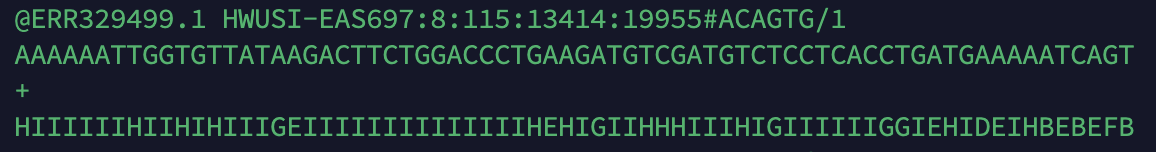
(二)fastq格式(*.fq)
通常是核酸序列以及测序质量得分情况
第一行:@开头,是序列的标识符以及描述信息
第二行:序列信息
第三行:+号
第四行:碱基质量值,与第二行对应,长度相当
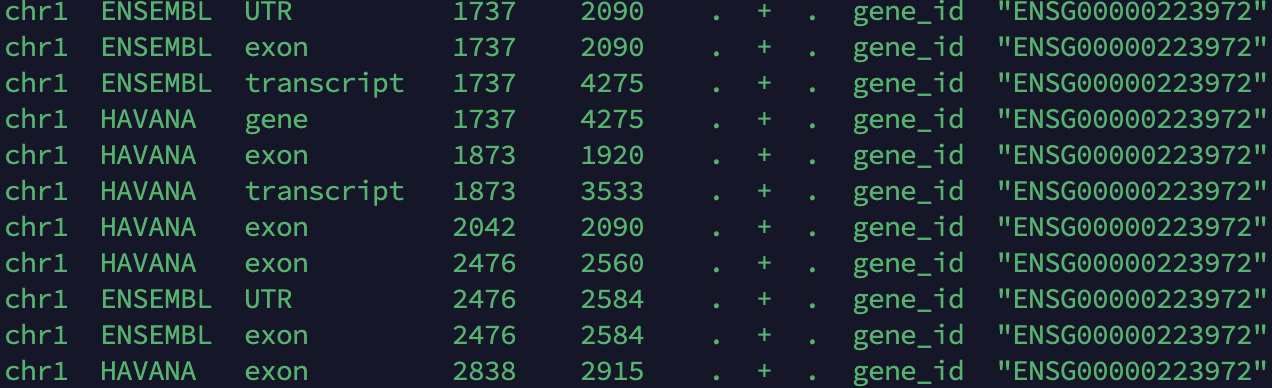
(三)gtt基因组注释文件

(四)gtf基因注释文件

二、文本处理工具——grep
1)常见参数:
-w:精确查找某个关键词
-c:统计成功匹配的行数
-v:反向选择,输出没有匹配的行
-n:显示匹配成功的行号
-r:在整个目录进行匹配 ??在这里目录必须和指令放在一起 eg:grep "gene" -r Data/ (-r和目录必须相连)
-e:可以指定多个匹配模式 eg: grep -e "word_1" -e "word_2" example.gtf
-f:从指定文件进行读取
-i:忽略大小写
$ less -S example.gtf | grep -w "gene" | less -S # 在example.gtf中准确匹配gene这个单词
chr1 HAVANA gene 1737 4275 . + . gene_id "ENSG00000223972"
chr1 HAVANA gene 4226 19433 . - . gene_id "ENSG00000227232"
chr1 HAVANA gene 19417 20972 . + . gene_id "ENSG00000243485"
chr1 ENSEMBL gene 20229 20366 . + . gene_id "ENSG00000221311"
chr1 HAVANA gene 24417 25944 . - . gene_id "ENSG00000237613"
chr1 ENSEMBL gene 42912 44799 . + . gene_id "ENSG00000233004"
chr1 HAVANA gene 52811 53750 . + . gene_id "ENSG00000240361"2)正则表达式

eg:grep '^T' #匹配行首的T
grep ')$' #匹配行尾)
grep '[^Tt]' #排除Tt
grep '[ATCG]' #匹配任意一个??grep使用小技巧
1)匹配不准确时可以延长匹配内容,增加匹配的限制
2)匹配之前可以先过滤,例如grep -v 先筛选一些
三、文本处理工具——sed
1)常用参数

2)用法
sed [-options] 'script' file ;其中:script=位置+操作
位置参数

操作参数

$ cat readme.txt | sed '1a Welcome to Biotrainee()'
Welcome to Biotrainee() !
Welcome to Biotrainee() #在第一行下面加上了一行
This is your personal account in our Cloud.
Have a fun with it.
Please feel free to contact with me( email to jmzeng1314@163.com )
(http://www.biotrainee.com/thread-1376-1-1.html)??改变字符的注意事项
$ cat readme.txt | sed 's/is/IS/'
Welcome to Biotrainee() !
ThIS is your personal account in our Cloud. #可以发现只有这一个is改变了
Have a fun with it.
Please feel free to contact with me( email to jmzeng1314@163.com )
(http://www.biotrainee.com/thread-1376-1-1.html)
#全局改变可以加上在's///g'
$ cat readme.txt | sed 's/is/IS/g' #这样子就可以改变全局啦!这个方法可以联系上一篇文章vim编辑器改变字符串
Welcome to Biotrainee() !
ThIS IS your personal account in our Cloud.
Have a fun with it.
Please feel free to contact with me( email to jmzeng1314@163.com )
(http://www.biotrainee.com/thread-1376-1-1.html)
#也可以指选择某一行改变
$ cat readme.txt | sed '4s/ee/EE/'
Welcome to Biotrainee() !
This is your personal account in our Cloud.
Have a fun with it.
Please fEEl free to contact with me( email to jmzeng1314@163.com ) #发现第四行只改变了一个
(http://www.biotrainee.com/thread-1376-1-1.html)
#末尾加上g,也可以改变一整列
$ cat readme.txt | sed '4s/ee/EE/g'
Welcome to Biotrainee() !
This is your personal account in our Cloud.
Have a fun with it.
Please fEEl frEE to contact with me( email to jmzeng1314@163.com )
(http://www.biotrainee.com/thread-1376-1-1.html)
????思考题
1)大小写转换 tr
(小写变大写) sed 's/[a-z]/\U&/g' ; tr a-z A-Z ; tr [:lower:][:upper:]
(大写变小写) sed 's/[A-Z]/\L&/g' ; tr A-Z a-z ; tr [:upper:][:lower:]
2)截取一行的前几个字符 cut
cut -c-num
$ cat readme.txt | cut -c-4 #截取每行的前4个字符
Welc
This
Have
Plea
(htt3)提取奇数/偶数行 sed
#提取奇数行
$ cat readme.txt | sed -n '1~2p'
Welcome to Biotrainee() !
Have a fun with it.
(http://www.biotrainee.com/thread-1376-1-1.html)
#提取偶数行
$ cat readme.txt | sed -n '0~2p'
This is your personal account in our Cloud.
Please feel free to contact with me( email to jmzeng1314@163.com )四、文本处理工具——awk
1)基本结构 :

2) 划分字段
$ 0 代表整个文本
$ 1 代表第一个数据字段
...
$ NF代表最后一个数据字段
注意??:awk默认分隔符是任意空白字符(如空格或tab键),也可以用-F来自定义分隔符。
#基础结构
$ less -S Data/example.gtf | awk '{print $9}' #按空格划分,取出第九个字段
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
gene_id
#匹配结构(先匹配UTR,再输出全部)
$ less -S Data/example.gtf | awk '/UTR/{print $0}' |less -S
#扩展结构(相当于开头和结尾添加文字)
$ less -S Data/example.gtf | awk 'BEGIN{print "find UTR feature" } /UTR/{print $0} END{print "end"}' |less -S
find UTR feature
chr1 ENSEMBL UTR 1737 2090 . + . gene_id "ENSG00000223972"; transcript_i
chr1 ENSEMBL UTR 2476 2584 . + . gene_id "ENSG00000223972"; transcript_i
chr1 ENSEMBL UTR 3084 4021 . + . gene_id "ENSG00000223972"; transcript_i
chr1 ENSEMBL UTR 4226 4561 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 4226 4692 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 4250 4275 . + . gene_id "ENSG00000223972"; transcript_i
chr1 ENSEMBL UTR 4833 4901 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 5659 5810 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 6470 6628 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 6721 6918 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 7096 7141 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 7414 7416 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 8227 8229 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 8869 8938 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 14601 14754 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 14601 14754 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 14707 14754 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 19184 19206 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 19184 19233 . - . gene_id "ENSG00000227232"; transcript_i
chr1 ENSEMBL UTR 19184 19233 . - . gene_id "ENSG00000227232"; transcript_i
chr1 HAVANA UTR 24417 25003 . - . gene_id "ENSG00000237613"; transcript_i
chr1 HAVANA UTR 25600 25944 . - . gene_id "ENSG00000237613"; transcript_i
chr1 ENSEMBL UTR 44797 44799 . + . gene_id "ENSG00000233004"; transcript_i
chr1 HAVANA UTR 58918 58953 . + . gene_id "ENSG00000177693"; transcript_i
chr1 HAVANA UTR 59869 59971 . + . gene_id "ENSG00000177693"; transcript_i
chr1 ENSEMBL UTR 127146 127148 . - . gene_id "ENSG00000237683"; transcript_i
chr2 HAVANA UTR 28814 31610 . - . gene_id "ENSG00000184731"; transcript_i
end3)内置参数

$ cat example.gtf | awk 'BEGIN{OFS=":"} {print $3,$4,$5}' |head -5
UTR:1737:2090
exon:1737:2090
transcript:1737:4275
gene:1737:4275
exon:1873:19204)awk 条件和循环语句
条件判断: awk '{ if (判断条件) {yes} else {no}' }
循环语句: awk ‘{ for {循环条件} {循环语句} }'
$ less -S Data/example.gtf | awk '{ if($3=="gene") print $0 }'| less -S #显示出第三列为gene的行
chr1 HAVANA gene 1737 4275 . + . gene_id "ENSG00000223972"; transcript_i
chr1 HAVANA gene 4226 19433 . - . gene_id "ENSG00000227232"; transcript_i
chr1 HAVANA gene 19417 20972 . + . gene_id "ENSG00000243485"; transcript_i
chr1 ENSEMBL gene 20229 20366 . + . gene_id "ENSG00000221311"; transcript_i
chr1 HAVANA gene 24417 25944 . - . gene_id "ENSG00000237613"; transcript_i
chr1 ENSEMBL gene 42912 44799 . + . gene_id "ENSG00000233004"; transcript_i
chr1 HAVANA gene 52811 53750 . + . gene_id "ENSG00000240361"; transcript_i5)算术运算

$ less -S example.gtf | awk '/exon/{print $5-$4}' | less -S
353
47
48
84
108
77
153
1191
217
466
466原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。