沉浸式体验WGBS(上游)

沉浸式体验WGBS(上游)

沉浸式体验WGBS(上游)

甲基化芯片数据处理我是有视频课程的
首先需要阅读我在生信技能树的甲基化系列教程,目录如下:
- 01-甲基化的一些基础知识.pdf
- 02-甲基化芯片的一般分析流程.pdf
- 03-甲基化芯片数据下载的多种技巧.pdf
- 04-甲基化芯片数据下载如何读入到R里面.pdf
- 05-甲基化芯片数据的一些质控指标.pdf
- 06-甲基化信号值矩阵差异分析哪家强.pdf
- 07-甲基化芯片信号值矩阵差异分析的标准代码.pdf
- 08-TCGA数据库的各个癌症甲基化芯片数据重新分析.pdf
- 09-TCGA数据库的癌症甲基化芯片数据重分析.pdf
- 10-TCGA数据辅助甲基化区域的功能研究.pdf
- 11-按基因在染色体上的顺序画差异甲基化热图.pdf
- 850K甲基化芯片数据的分析.pdf
- 使用DSS包多种方式检验差异甲基化信号区域.pdf
然后就可以看我在B站免费分享的视频课程《甲基化芯片(450K或者850K)数据处理 》
- 教学视频免费在:https://www.bilibili.com/video/BV177411U7oj
- 课程配套思维导图:https://mubu.com/doc/1cwlFgcXMg
但是另外两个高频甲基化技术我却一直没有机会涉及,就是WGBS和RRBS,恰好学徒整理了这方面知识点, 借花献佛给大家。
0. 知识点
最高频的3个甲基化技术
这3个甲基化技术就是 甲基化测序的 WGBS和RRBS,还有 芯片:
- 全基因组DNA甲基化测序(Whole Genome Bisulfite Sequencing,WGBS)是 DNA 甲基化研究的金标准,它通过 Bisulfite 处理和全基因组 DNA 测序结合的方式,对整个基因组上的甲基化情况进行分析,具有单碱基分辨率,可精确评估单个 C 碱基的甲基化水平,构建全基因组精细甲基化图谱。数据量非常大。
- 简化甲基化测序 (Reduced representation bisulfite sequencing, RRBS)是一种准确、高效、经济的DNA甲基化研究方法,通过酶切 (Msp I) 富集启动子及CpG岛区域,并进行Bisulfite测序,同时实现DNA甲基化状态检测的高分辨率和测序数据的高利用率。作为一种高性价比的甲基化研究方法,简化甲基化测序在大规模临床样本的研究中具有广泛的应用前景。
- Illumina的Infinium BeadChip芯片,包括HumanMethyation450(450K)和MethylationEPIC(850K)。Infinium芯片存在染料偏差、不同探针化学和位置效应的问题,已知这些问题会影响结果,必须在数据处理过程中进行校正。Infinium 450K探针交叉反应和模糊比对到人类基因组中的多个位置影响了485,000个探测器中的约140,000个探针(29%),将可用探针的数量减少到约345,000个。这个问题在新发布850K仍然存在,其包括> 90%的450K探针。
三种类型(CpG/CHG/CHH)
在bismark中,根据甲基化的C所处的上下文环境,分成以下3类;
- CpG
- CHG
- CHH
p代表磷酸二酯键,CpG指的是甲基化的C的下游是1个G碱基
H代表除了G碱基之外的其他碱基,即A, C, T中的任意一种
CHG代表甲基化的C下游的2个碱基是H和G, CHH表示甲基化的C下游的两个碱基都是H
亚硫酸氢盐测序(BS-seq)
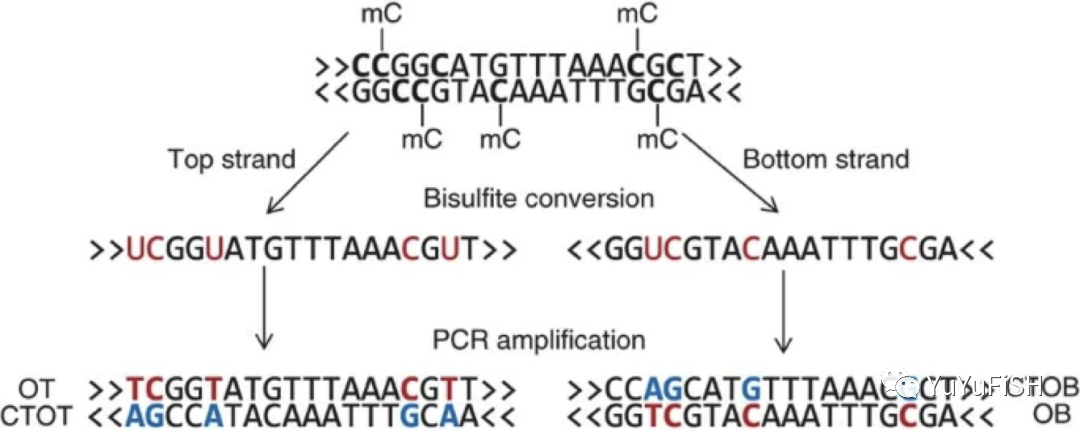
样本用 Bisulfite 处理,将基因组中未发生甲基化的 C 碱基转换成 U,而甲基化的C则保持不变,进行PCR扩增后变成T,与原本具有甲基化修饰的 C 碱基区分开来,再结合高通量测序技术,与参考序列比对。
- 未甲基化的 C -> T
- 甲基化的 C -> C
对 PCR产物进行高通量测序,再与C->T和G->A的参考基因组序列比对确定唯一最佳比对后,最后与正常的参考基因组进行对比,即可判断CpG/CHG/CHH context的C是否发生了甲基化。
分析步骤
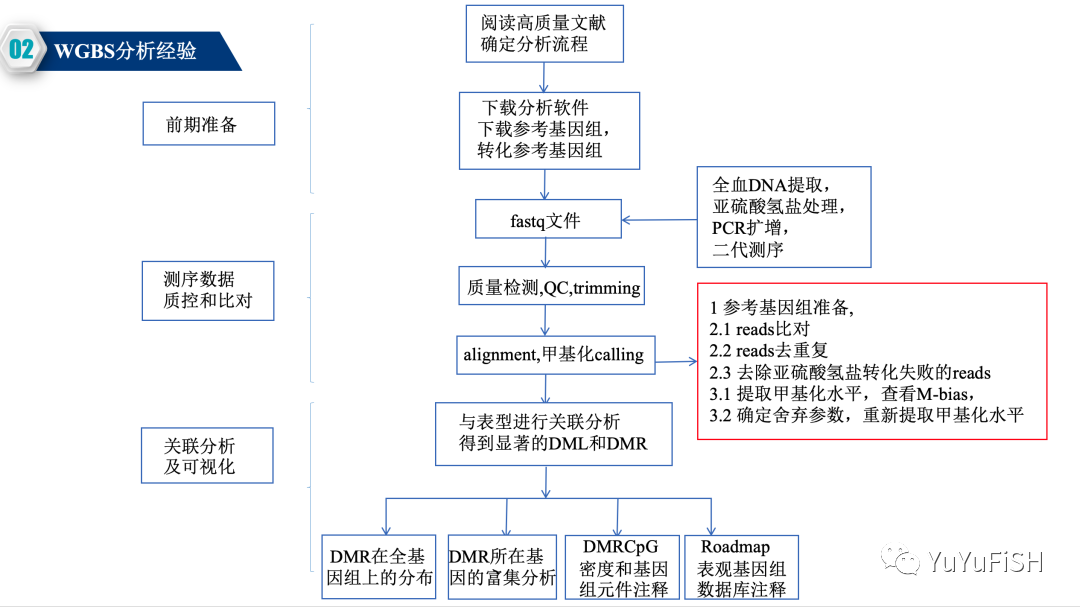
质控,过滤:参考转录组的步骤
从比对开始就是WGBS上游分析重点:Bismark软件
下面是针对不同甲基化技术,Bismark步骤的变化
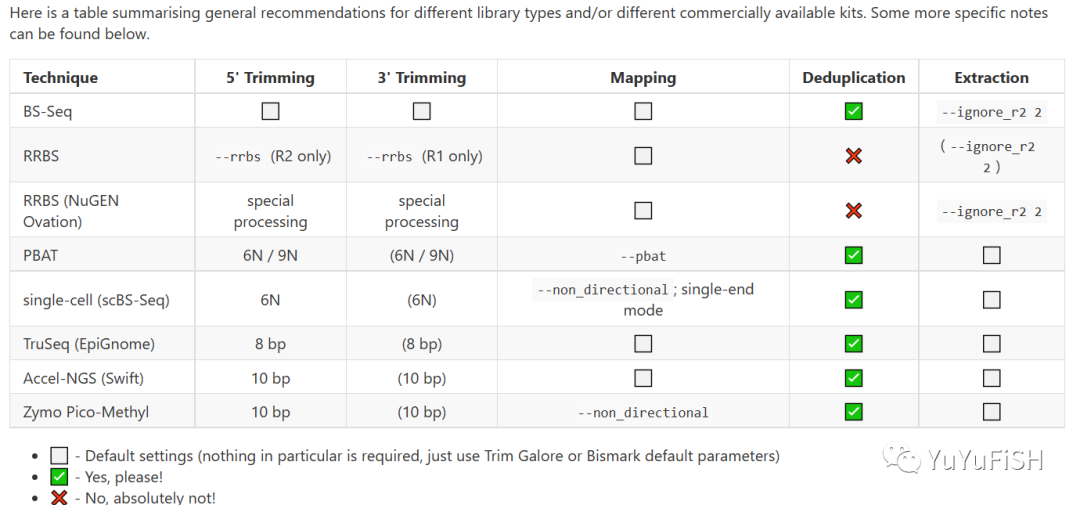
例如,在去重复这一步WGBS需要做,RRBS一定不要
开始走一趟,分析WGBS数据~~~
1. 安装软件
1.1 新建小环境
## conda管理环境
# 创建名为snakemake的软件环境来安装转录组学分析的生物信息学软件
# 创建小环境成功,并成功安装python3版本,每建立一个小环境,安装一个python=3的软件作为依赖
conda create -y -n snakemake python=3
# 查看当前conda环境
conda info --e
# 激活
conda activate snakemake
1.2 示范下载Bismark软件
下载了软件之后,会在家目录产生Bismark文件夹,里面放了函数还有说明书,可以下载到本地查看
说明书:
Bismark/Docs at master · FelixKrueger/Bismark · GitHub
Bismark (felixkrueger.github.io)
(snakemake) yulan 23:35:37 ~
$ git clone https://github.com/FelixKrueger/Bismark.git
Cloning into 'Bismark'...
remote: Enumerating objects: 3772, done.
remote: Counting objects: 100% (616/616), done.
remote: Compressing objects: 100% (288/288), done.
remote: Total 3772 (delta 341), reused 543 (delta 285), pack-reused 3156
Receiving objects: 100% (3772/3772), 38.94 MiB | 14.48 MiB/s, done.
Resolving deltas: 100% (2375/2375), done.
(snakemake) yulan 23:37:24 ~
$ cd Bismark/
(snakemake) yulan 23:37:45 ~/Bismark
$ ls
bam2nuc Bismark_alignment_modes.pdf copy_bismark_files_for_release.pl license.txt test_data.fastq
bismark bismark_genome_preparation coverage2cytosine methylation_consistency travis_files
bismark2bedGraph bismark_methylation_extractor deduplicate_bismark NOMe_filtering
bismark2report CHANGELOG.md Docs plotly
bismark2summary _config.yml filter_non_conversion README.md
(snakemake) yulan 23:37:47 ~/Bismark
$ ./bismark_genome_preparation -version
Bismark - Bisulfite Mapper and Methylation Caller.
Bismark Genome Preparation Version: v0.24.0
Copyright 2010-22, Felix Krueger, Altos Bioinformatics
https://github.com/FelixKrueger/Bismark
(snakemake) yulan 23:50:16 ~/Bismark
$ pwd
/home/yulan/Bismark
Notes:需要用户提前安装好以下软件:
1.Bowtie2(http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) 2.Hisat2(https://ccb.jhu.edu/software/hisat2/index.shtml) 3.Samtools(http://samtools.sourceforge.net/)
1.3 输入到环境变量中
# 查看环境变量
$ echo $PATH|tr ':' '\n'
/home/yulan/bin
/home/yulan/.sdkman/candidates/java/current/bin
/home/yulan/miniconda3/envs/snakemake/bin
/home/yulan/miniconda3/condabin
/usr/local/sbin
/usr/local/bin
/usr/sbin
/usr/bin
/sbin
/bin
/usr/games
/usr/local/games
/snap/bin
# 将这个文件夹的路径添加到环境变量 $PATH 中,同时写入到 .bashrc
echo "PATH=${HOME}/Bismark:$PATH" >> ${HOME}/.bashrc
# 重新激活一下环境,或者重新登录一下服务器
source ~/.bashrc
下面例子均使用hisat2做示范
1.4 创建目录
使用 mkdir 创建多个文件夹存放数据
yulan 14:50:14 ~/wgbs_test
$ tree -L 1
.
├── cleandata # 质控后的数据
├── duplicate # reads的去重复
├── extractor #提取DNA甲基化
├── mapping #储存mapping结果数据的文件夹
├── nohup.out
├── qc
├── rawdata #原始数据
└── temp_bismark #储存中间数据的文件夹
7 directories, 1 file
2. 数据前期处理
2.1 aspera下载fastq数据
系统性红斑狼疮数据集(双端测序,2021)
- 数据集:
GSE146410 :GSM4384986样本
PRJNA610526
- 作者的处理手法:

首先去数据库,默认选择列,下载原始数据的基本信息:ENA Browser (ebi.ac.uk)
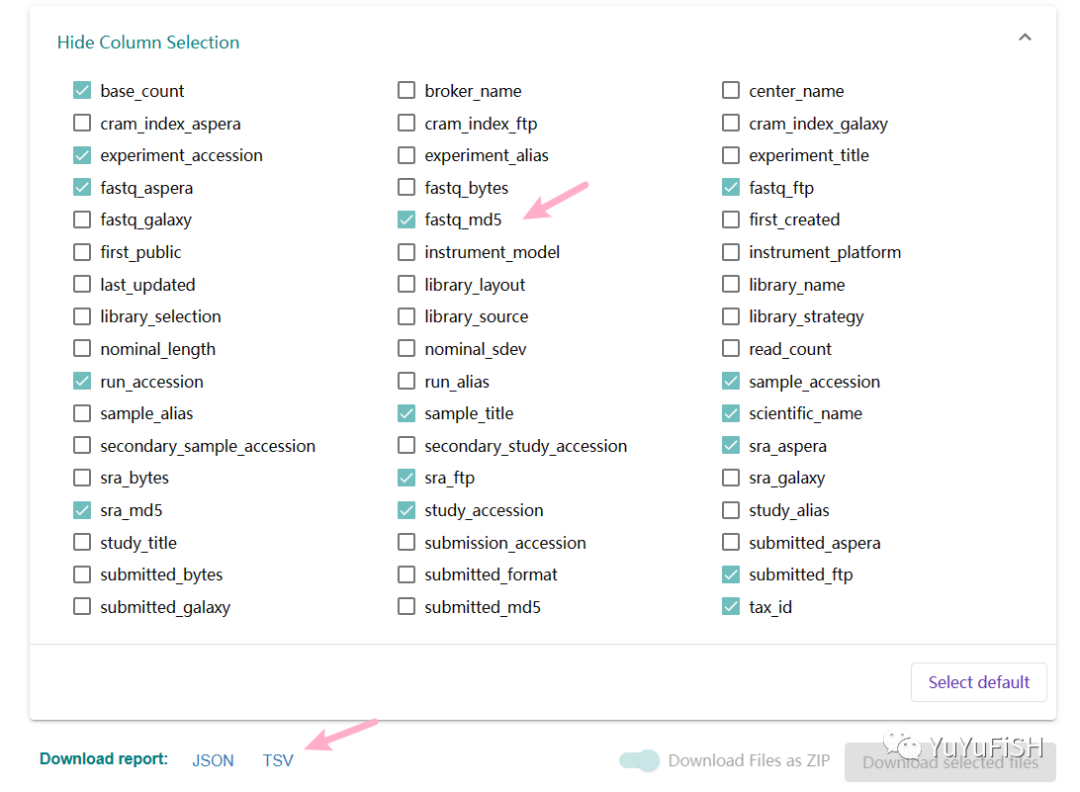
# 因为在小环境snakemake下运行,选择第二个密钥
(snakemake) yulan 14:55:14 ~
$ find ./ -name asperaweb_id_dsa.openssh
./miniconda3/envs/rna/etc/asperaweb_id_dsa.openssh
./miniconda3/envs/snakemake/etc/asperaweb_id_dsa.openssh
./miniconda3/pkgs/aspera-cli-3.9.6-h5e1937b_0/etc/asperaweb_id_dsa.openssh
# 进入文件夹
cd rawdata/
# 下载单个文件
# sra格式
ascp -k 1 -QT -l 500m -P33001 -i /home/yulan/miniconda3/envs/snakemake/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/[vol1/fastq/SRR112/055/SRR11243555/SRR11243555_1.fastq.gz](ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR112/055/SRR11243555/SRR11243555_1.fastq.gz) .
ascp -k 1 -QT -l 500m -P33001 -i /home/yulan/miniconda3/envs/snakemake/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/[vol1/fastq/SRR112/055/SRR11243555/SRR11243555_2.fastq.gz](ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR112/055/SRR11243555/SRR11243555_2.fastq.gz) .
##注意
#寻找密钥:asperaweb_id_dsa.openssh是密钥地址,每次都要寻找!
#方法:转成家目录~,在当前目录下寻找输入:find ./ -name asperaweb_id_dsa.openssh
#前面那一段有miniconda3是要换的,换自己miniconda3的地址
#后面era那个是看filereport_read_run_PRJNA610526_tsv.txt里面的,
#最后的.意思是下载到当前目录
##########################################################

2.2 md5值检验
数据完整性检验(非常重要!!!),防止下载不完全
# 查看文件
(snakemake) yulan 17:55:12 ~/wgbs_test/rawdata
$ less filereport_read_run_PRJNA610526_tsv.txt | column -t | less -S
## 数据完整性检验(非常重要!!!)
# 得到md5值
# 分号隔开了两个fastq文件的md5值
(snakemake) yulan 18:03:35 ~/wgbs_test/rawdata
$ awk 'NR>2{print $9"\t"$4}' filereport_read_run_PRJNA610526_tsv.txt
8c022680c5af055c0784d63d07d511ed;24eae505238fc1b10e151697057e54bc SRR11243555
# 手动写进md5.txt
(snakemake) yulan 15:03:16 ~/wgbs_test/rawdata
$ cat md5.txt
8c022680c5af055c0784d63d07d511ed SRR11243555_1.fastq.gz
24eae505238fc1b10e151697057e54bc SRR11243555_2.fastq.gz
# md5值检验
(snakemake) yulan 15:03:25 ~/wgbs_test/rawdata
$ md5sum -c md5.txt
SRR11243555_1.fastq.gz: OK
SRR11243555_2.fastq.gz: OK
2.3 fastqc质控
# 使用FastQC软件对单个fastq文件进行质量评估,结果输出到qc/文件夹下
qcdir=/home/yulan/wgbs_test/qc
fqdir=/home/yulan/wgbs_test/rawdata
# 多个数据质控
fastqc -t 2 -o $qcdir $fqdir/SRR*.fastq.gz
(snakemake) yulan 19:52:57 ~/wgbs_test/qc
$ ll
total 1804
drwxrwxr-x 2 yulan yulan 4096 1月 5 17:37 ./
drwxrwxr-x 4 yulan yulan 4096 1月 5 17:06 ../
-rw-rw-r-- 1 yulan yulan 598572 1月 5 17:37 SRR11243555_1_fastqc.html
-rw-rw-r-- 1 yulan yulan 310486 1月 5 17:37 SRR11243555_1_fastqc.zip
-rw-rw-r-- 1 yulan yulan 605256 1月 5 17:37 SRR11243555_2_fastqc.html
-rw-rw-r-- 1 yulan yulan 318028 1月 5 17:37 SRR11243555_2_fastqc.zip
(snakemake) yulan 19:52:58 ~/wgbs_test/qc
$ multiqc *.zip
/// MultiQC ? | v1.13.dev0
| multiqc | Search path : /home/data/yulan/wgbs_test/qc/SRR11243555_1_fastqc.zip
| multiqc | Search path : /home/data/yulan/wgbs_test/qc/SRR11243555_2_fastqc.zip
| searching | ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 2/2
| fastqc | Found 2 reports
| multiqc | Compressing plot data
| multiqc | Report : multiqc_report.html
| multiqc | Data : multiqc_data
| multiqc | MultiQC complete
2.4 trim_galore过滤
(snakemake) yulan 20:11:08 ~/wgbs_test/rawdata
$ cat sampleId.txt
SRR11243555
# 定义文件夹
rawdata=/home/yulan/wgbs_test/rawdata
cleandata=/home/yulan/wgbs_test/cleandata/trim_galore
# 多个
cat /home/yulan/wgbs_test/rawdata/sampleId.txt | while read id
do
echo "trim_galore --phred33 -j 5 -q 20 --length 36 --stringency 3 --fastqc --paired --max_n 3 -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz"
done >trim_galore.sh
##注意
##上面的参数需要根据自己实际项目修改,参数可参考ppt
##自己需要创建sampleID.txt,tounch创建,vim里面输入SRR
##上面长篇代码的规律输出成下面截图的样子,无疑就是拼接
nohup sh trim_galore.sh >trim_galore.log &
#保留处理日志的习惯;cat trim_galore.log可看下有没有处理完成
#结果SRR1039508_1.fastq.gz_trimming_report.txt存放处理的信息
# 使用MultiQc整合FastQC结果
multiqc *.zip
- 结果
yulan 23:06:42 ~/wgbs_test/cleandata/trim_galore
$ ll -h
total 34G
drwxrwxr-x 2 yulan yulan 4.0K 1月 5 22:59 ./
drwxrwxr-x 3 yulan yulan 4.0K 1月 5 20:06 ../
-rw-rw-r-- 1 yulan yulan 5.3K 1月 5 20:51 SRR11243555_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 yulan yulan 587K 1月 5 22:30 SRR11243555_1_val_1_fastqc.html
-rw-rw-r-- 1 yulan yulan 306K 1月 5 22:30 SRR11243555_1_val_1_fastqc.zip
-rw-rw-r-- 1 yulan yulan 18G 1月 5 22:01 SRR11243555_1_val_1.fq.gz
-rw-rw-r-- 1 yulan yulan 5.4K 1月 5 22:01 SRR11243555_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 yulan yulan 589K 1月 5 22:59 SRR11243555_2_val_2_fastqc.html
-rw-rw-r-- 1 yulan yulan 309K 1月 5 22:59 SRR11243555_2_val_2_fastqc.zip
-rw-rw-r-- 1 yulan yulan 17G 1月 5 22:01 SRR11243555_2_val_2.fq.gz
-rw-rw-r-- 1 yulan yulan 18K 1月 5 22:59 trim_galore.log
-rw-rw-r-- 1 yulan yulan 241 1月 5 20:16 trim_galore.sh
# 查看过滤后质控结果
yulan 23:06:46 ~/wgbs_test/cleandata/trim_galore
$ multiqc *.zip
3. mapping 比对???
3.1 准备参考基因组
之前学习转录组上游时已经下载了参考基因组GRCh38.106
请先下载参考基因组并将其放在基因组文件夹中,基因组可以 从Ensembl(http://www.ensembl.org/info/data/ftp/index.html/) 或NCBI网站 (ftp://ftp.ncbi.nih.gov/genomes/)下载。Bismark 支持 FastA 格式的参考基因组序列文件,允许文件扩展名是 .fa或 .fasta。
3.2 转化参考基因组
接着使用bismark_genome_preparation转化参考基因组,会生成C->T 和 G->A 版本的基因组;
你需要指定参考基因组的目录,其中要包含比对需要读取的基因组(需要当前此文件夹中的 FastA 文件,扩展名为 .fa 或 .fasta,每个文件有单个或多个序列)。 Bismark 将在此目录中创建两个单独的文件夹,一个用于 C->T 转换的基因组,另一个用于 G->A 转换的基因组。 创建 C->T 和 G->A 版本的基因组后,它们将使用 bowtie-build (或 bowtie2-build) 并行索引。一旦创建了 C->T 和 G->A 基因组索引,就不需要再次使用该脚本(除非你想比对不同的基因组)。 请注意,Bowtie 1 和 Bowtie 2 索引不兼容。要创建用于 Bowtie 2 的基因组索引,还需要包含选项 --bowtie2。
# 搜索hisat2安装位置
(snakemake) yulan 23:43:16 ~
$ which hisat2
/home/yulan/miniconda3/envs/snakemake/bin/hisat2
# 别人下好的基因组
(snakemake) yulan 23:49:07 ~
$ ll /home/junliu/database/GRCh38.106/
total 4423588
drwxrwxr-x 3 junliu junliu 4096 7月 8 09:47 ./
drwxrwxr-x 4 junliu junliu 4096 7月 8 09:42 ../
drwxrwxr-x 2 junliu junliu 4096 7月 8 10:04 Hisat2Index/
-rw-rw-r-- 1 junliu junliu 1378262703 7月 8 09:27 Homo_sapiens.GRCh38.106.chr.gtf
-rw-rw-r-- 1 junliu junliu 3151425857 7月 8 09:28 Homo_sapiens.GRCh38.dna.primary_assembly.fa
-rw------- 1 junliu junliu 36688 7月 8 10:06 nohup.out
# 软连接别人的参考基因组
ln -s /home/junliu/database/GRCh38.106/Homo_sapiens.GRCh38.dna.primary_assembly.fa database/hg38
#这一步比较耗时,人类参考基因组耗时>1h
nohup bismark_genome_preparation --hisat2 --path_to_aligner /home/yulan/miniconda3/envs/snakemake/bin --verbose /home/yulan/database/hg38 &
# 重要参数说明
bismark_genome_preparation
--bowtie2/--hisat2:调用bowtie2/hisat2建立基因组索引
--path_to_aligner:比对软件所在文件夹的全路径
--verbose:输出详情(nohup.out)
--parallel:设置线程,索引建立是并行运行,因此实际线程要×2
--large-index:大基因组索引建立
--yes:如果有安全类问题则自动选择yes,比如覆盖某个已存在的文件
<path_to_genome_folder>:基因组所在文件夹路径,即~/bismark_example/01index/
- 结果
- 构建完成后会在/home/yulan/database/hg38下生成一个Bisulfite_Genome文件夹,存放CT_conversion和GA_conversion的转化后参考基因组。
(snakemake) yulan 11:54:33 ~/wgbs_test/mapping
$ ll -h /home/yulan/database/hg38/
total 16K
drwxrwxr-x 3 yulan yulan 4.0K 1月 5 11:27 ./
drwxrwxr-x 3 yulan yulan 4.0K 1月 5 10:52 ../
drwxrwxr-x 4 yulan yulan 4.0K 1月 5 11:27 Bisulfite_Genome/
lrwxrwxrwx 1 yulan yulan 76 1月 5 10:53 Homo_sapiens.GRCh38.dna.primary_assembly.fa -> /home/junliu/database/GRCh38.106/Homo_sapiens.GRCh38.dna.primary_assembly.fa
(snakemake) yulan 13:44:18 ~/wgbs_test
$ ll /home/yulan/database/hg38/Bisulfite_Genome/
total 16
drwxrwxr-x 4 yulan yulan 4096 1月 5 11:27 ./
drwxrwxr-x 3 yulan yulan 4096 1月 5 11:27 ../
drwxrwxr-x 2 yulan yulan 4096 1月 5 12:32 CT_conversion/
drwxrwxr-x 2 yulan yulan 4096 1月 5 12:29 GA_conversion/
(snakemake) yulan 13:41:36 ~/wgbs_test
$ tree /home/yulan/database/hg38/Bisulfite_Genome/
/home/yulan/database/hg38/Bisulfite_Genome/
├── CT_conversion
│ ├── BS_CT.1.ht2
│ ├── BS_CT.2.ht2
│ ├── BS_CT.3.ht2
│ ├── BS_CT.4.ht2
│ ├── BS_CT.5.ht2
│ ├── BS_CT.6.ht2
│ ├── BS_CT.7.ht2
│ ├── BS_CT.8.ht2
│ └── genome_mfa.CT_conversion.fa
└── GA_conversion
├── BS_GA.1.ht2
├── BS_GA.2.ht2
├── BS_GA.3.ht2
├── BS_GA.4.ht2
├── BS_GA.5.ht2
├── BS_GA.6.ht2
├── BS_GA.7.ht2
├── BS_GA.8.ht2
└── genome_mfa.GA_conversion.fa
2 directories, 18 files
3.3 reads比对 (非常慢)
此步骤代表实际的亚硫酸氢盐比对和甲基化calling部分。Bismark要求指定两个文件:
- 1.包含参考基因组的目录。此文件夹必须包含未修改的基因组(如 .fa 或 .fasta 文件)以及在 Bismark 基因组准备步骤中生成的两个亚硫酸氢盐基因组子目录。
- 2.要分析的序列文件(FastQ 或 FastA 格式)
- 3. 根据自己电脑配置加线程
- 4. 示例使用示例双端测序
先解压SRR11243555_1_val_1.fq.gz 成 fq 模式(脑子一热,给他解压掉了,其实可以直接用压缩文件gz分析)
(snakemake)yulan 23:18:09 ~/wgbs_test/cleandata/trim_galore
$ gzip -d SRR11243555_1_val_1.fq.gz
(snakemake)yulan 23:18:09 ~/wgbs_test/cleandata/trim_galore
$ gzip -d SRR11243555_2_val_2.fq.gz
代码:
(snakemake)yulan 23:45:29 ~/wgbs_test/mapping
$ pwd
/home/yulan/wgbs_test/mapping
(snakemake)yulan 23:45:31 ~/wgbs_test/mapping
$ cat >mapping.sh
bismark --hisat2 --genome /home/yulan/database/hg38 -X 200 --temp_dir /home/yulan/wgbs_test/temp_bismark/ -o /home/yulan/wgbs_test/mapping/ -B Testpaired -1 /home/yulan/wgbs_test/cleandata/trim_galore/SRR11243555_1_val_1.fq -2 /home/yulan/wgbs_test/cleandata/trim_galore/SRR11243555_2_val_2.fq
^C
#结果位于/home/yulan/wgbs_test/mapping/
(snakemake)yulan 23:45:33 ~/wgbs_test/mapping
$ nohup bash mapping.sh >mapping.log &
# 重要参数说明
bismark
-N:设置seed中允许的最大错配数,可取0或1,默认为0。值越高,对齐速度越慢,灵敏度越高。
-L:设置seed长度,最大值为32,默认为20。值越高,对齐速度越快,灵敏度越低。
--quiet:不输出比对流程信息
--un:过滤多处匹配的reads
--ambiguous:多处匹配reads信息独立记录
--sam/--bam:输出SAM格式,与--parallel不兼容/输出BAM格式,可调整--parallel
--bowtie2/--hisat2 :调用bowtie2/hisat2,默认bowtie2
--path_to_bowtie2/--path_to_hisat2 :bowtie2/hisat2所在文件夹的全路径
-o/--output_dir :输出文件的全路径
--samtools_path:samtools所在文件夹的全路径
--prefix:指定输出文件的前缀
--q/--fastq:输入文件为FastQ
-f/--fasta:输入文件为FastA
--phred33-quals/--phred64-quals:指定FastQ文件的质量分数格式,默认为phred33
--genome:包含未修改的参考基因组和bismark_genome_preparation创建的子文件夹(CT_conversion/和GA_conversion/)的文件夹的路径,即~/bismark_example/01index/
-1/-2:双端测序文件
-X (最大插入片段长度,默认值:500)#衡量比对完成后的pair1和pair2最远可以相距的距离, 对于paired-end测序,即使一对reads成功比对到基因组上,但若它们相隔太远,Bismark则会丢弃它们。插入片段长度= reads1 length + gap length + reads 2 例如150-bp测序长度,reads1和reads2比对后最远可以相距300-bp,而X=300+150x2,这个参数需要根据建库后的DNA片段长度分布进行适当修改,因为X越大比对会越慢,但是X过小会导致丢失一部分reads;
与Bowtie2比对敏感性有关的参数为:
-L(seed长度)
-N(seed中最大允许的错配数)
--score_min (一个有效比对允许的最低得分),其默认值为L,0,-0.2,即f(x) = 0 + -0.2 * x(x是read长度)。对于一个75bp的read其被允许的最低比对得分为-15,这大概相当于在这条read中最多允许存在2个错配或者2个1-2bp的indel。详细参数可查看官网说明
# 输出文件
(a) Testpaired_pe.bam 所有对齐和甲基化的信息
(b) Testpaired_PE_report.txt 对齐和甲基化的主要信息概括
结果
- Testpaired_PE_report.txt 文件为结果概述
- Testpaired_pe.bam 文件储存了成功比对的reads信息
yulan 23:16:40 ~/wgbs_test/mapping
$ ll -h
total 49G
drwxrwxr-x 2 yulan yulan 4.0K 1月 6 00:21 ./
drwxrwxr-x 7 yulan yulan 4.0K 1月 5 23:11 ../
-rw-rw-r-- 1 yulan yulan 35K 1月 7 22:26 mapping.log
-rw-rw-r-- 1 yulan yulan 315 1月 6 11:17 mapping.sh
-rw-rw-r-- 1 yulan yulan 49G 1月 7 22:26 Testpaired_pe.bam #储存了成功比对的reads信息
-rw-rw-r-- 1 yulan yulan 2.0K 1月 7 22:26 Testpaired_PE_report.txt #结果概述
Testpaired_pe.bam:成功比对的reads
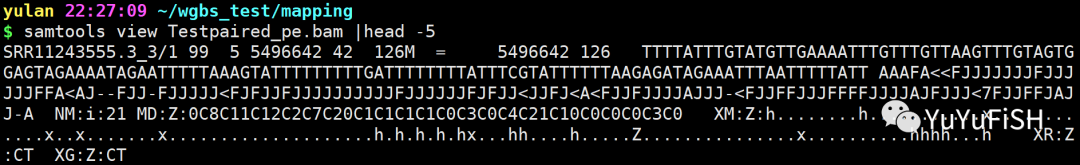
详细描述:
甲基化调用字符串包含一个点“.”代表 BS-read 中不涉及胞嘧啶的每个位置,或者包含以下三个不同胞嘧啶甲基化上下文的字母之一(大写 = 甲基化,小写 = 未甲基化):
字母 | 含义 |
|---|---|
z | unmethylated C in CpG context |
Z | methylated C in CpG context |
x | unmethylated C in CHG context |
X | methylated C in CHG context |
h | unmethylated C in CHH context |
H | methylated C in CHH context |
u | unmethylated C in Unknown context (CN or CHN) |
U | methylated C in Unknown context (CN or CHN) |
对于CpG, 采用字母Z的大小写来表征甲基化状态;对于CHG, 采用字母X的大小写来表征甲基化状态;对于CHH, 采用字母H 的大小写来表征甲基化状态。
Testpaired_PE_report.txt :结果描述
(snakemake) yulan 15:26:31 ~/wgbs_test/mapping
$ cat Testpaired_PE_report.txt
Bismark report for: /home/yulan/wgbs_test/cleandata/trim_galore/SRR11243555_1_val_1.fq and /home/yulan/wgbs_test/cleandata/trim_galore/SRR11243555_2_val_2.fq (version: v0.24.0)
Bismark was run with HISAT2 against the bisulfite genome of /home/data/yulan/database/hg38/ with the specified options: -q --score-min L,0,-0.2 --ignore-quals --no-mixed --no-discordant --maxins 200 --no-softclip --omit-sec-seq
Option '--directional' specified (default mode): alignments to complementary strands (CTOT, CTOB) were ignored (i.e. not performed)
Final Alignment report
======================
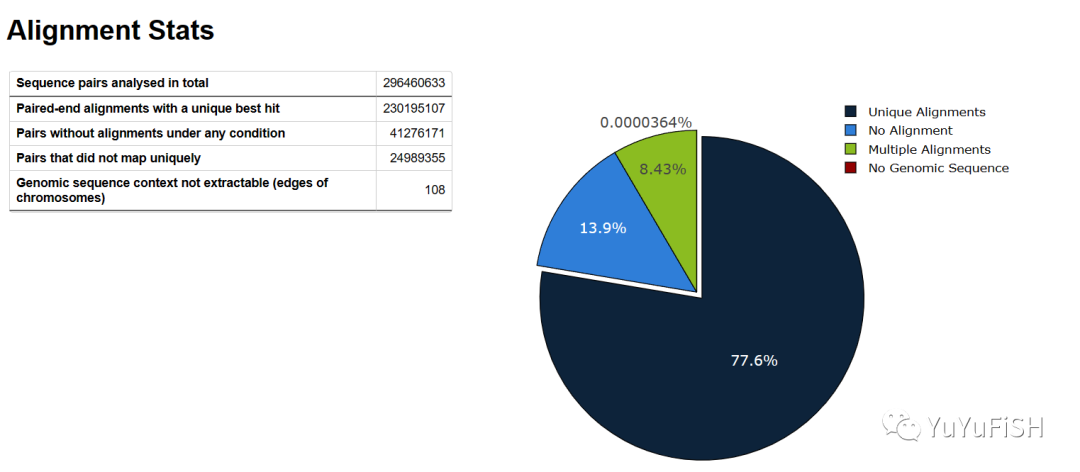
Sequence pairs analysed in total: 296460633
Number of paired-end alignments with a unique best hit: 230195107
Mapping efficiency: 77.6%
Sequence pairs with no alignments under any condition: 41276171
Sequence pairs did not map uniquely: 24989355
Sequence pairs which were discarded because genomic sequence could not be extracted: 108
Number of sequence pairs with unique best (first) alignment came from the bowtie output:
CT/GA/CT: 115948716 ((converted) top strand)
GA/CT/CT: 0 (complementary to (converted) top strand)
GA/CT/GA: 0 (complementary to (converted) bottom strand)
CT/GA/GA: 114246283 ((converted) bottom strand)
Number of alignments to (merely theoretical) complementary strands being rejected in total: 0
Final Cytosine Methylation Report
=================================
Total number of C's analysed: 10304205121
Total methylated C's in CpG context: 437990977
Total methylated C's in CHG context: 9684697
Total methylated C's in CHH context: 30760823
Total methylated C's in Unknown context: 22542
Total unmethylated C's in CpG context: 103555885
Total unmethylated C's in CHG context: 2359079347
Total unmethylated C's in CHH context: 7363133392
Total unmethylated C's in Unknown context: 2205065
C methylated in CpG context: 80.9%
C methylated in CHG context: 0.4%
C methylated in CHH context: 0.4%
C methylated in unknown context (CN or CHN): 1.0%
Bismark completed in 1d 11h 9m 10s
3.4 生成报告
直接运行bismark2report生成Testpaired_PE_report.html报告
(snakemake)yulan 18:38:57 ~/wgbs_test/mapping
$ bismark2report
Found 1 alignment reports in current directory. Now trying to figure out whether there are corresponding optional reports
Writing Bismark HTML report to >> Testpaired_PE_report.html <<
==============================================================================================================
Using the following alignment report: > Testpaired_PE_report.txt <
Processing alignment report Testpaired_PE_report.txt ...
Complete
No deduplication report file specified, skipping this step
No splitting report file specified, skipping this step
No M-bias report file specified, skipping this step
No nucleotide coverage report file specified, skipping this step
==============================================================================================================
(snakemake)yulan 18:39:00 ~/wgbs_test/mapping
$ ll -h
total 49G
drwxrwxr-x 2 yulan yulan 4.0K 1月 8 18:39 ./
drwxrwxr-x 9 yulan yulan 4.0K 1月 8 15:23 ../
-rw-rw-r-- 1 yulan yulan 35K 1月 7 22:26 mapping.log
-rw-rw-r-- 1 yulan yulan 315 1月 6 11:17 mapping.sh
-rw-rw-r-- 1 yulan yulan 49G 1月 7 22:26 Testpaired_pe.bam
-rw-rw-r-- 1 yulan yulan 3.1M 1月 8 18:39 Testpaired_PE_report.html
-rw-rw-r-- 1 yulan yulan 2.0K 1月 7 22:26 Testpaired_PE_report.txt
3.5 reads的去重复
去重复这一步WGBS需要做,RRBS一定不要
#去重复这一步WGBS需要做,RRBS一定不要
deduplicate_bismark --help
(snakemake)yulan 23:39:29 ~/wgbs_test/mapping
$ which samtools
/home/yulan/miniconda3/bin/samtools
#paired
deduplicate_bismark -p --bam /home/yulan/wgbs_test/mapping/Testpaired_pe.bam --samtools_path /home/yulan/miniconda3/bin --output_dir /home/yulan/wgbs_test/duplicate
nohup bash duplicate.sh >duplicate.log &
# 重要参数说明
deduplicate_bismark
--sam/--bam:删除 Bismark 对齐产生的SAM/BAM 文件中的重复数据,建议用于WGBS,但不建议应用于RRS (reduced representation shotgun),如 RRBS、amplicon or target enrichment libraries。
-p/--paired :前一步双端数据产生的结果文件
-s/--single:前一步单端数据产生的结果文件
--samtools_path:samtools所在文件夹的全路径
--output_dir:输出文件夹路径
--multiple:指定输入文件都作为一个样本处理,连接在一起进行重复数据删除。
对SAM文件使用Unix“cat”,对BAM文件使用“samtools cat”。所有输入文件的格式必须相同。默认情况下,标头取自要连接的第一个文件。
# 输出文件
(a) Testpaired_pe.deduplicated.bam
(b) Testpaired_pe.deduplication_report.txt
- 结果
Testpaired_pe.deduplication_report.txt:去重复后的结果描述
(snakemake)yulan 10:36:33 ~/wgbs_test/duplicate
$ ll -h
total 38G
drwxrwxr-x 2 yulan yulan 4.0K 1月 7 23:49 ./
drwxrwxr-x 8 yulan yulan 4.0K 1月 7 23:42 ../
-rw-rw-r-- 1 yulan yulan 13K 1月 8 00:40 duplicate.log
-rw-rw-r-- 1 yulan yulan 185 1月 7 23:49 duplicate.sh
-rw-rw-r-- 1 yulan yulan 38G 1月 8 00:39 Testpaired_pe.deduplicated.bam
-rw-rw-r-- 1 yulan yulan 307 1月 8 00:39 Testpaired_pe.deduplication_report.txt
(snakemake)yulan 10:36:37 ~/wgbs_test/duplicate
$ cat Testpaired_pe.deduplication_report.txt
Total number of alignments analysed in /home/yulan/wgbs_test/mapping/Testpaired_pe.bam: 230194999
Total number duplicated alignments removed: 53488617 (23.24%)
Duplicated alignments were found at: 39478415 different position(s)
Total count of deduplicated leftover sequences: 176706382 (76.76% of total)
3.6 去除重亚硫酸盐转化失败的reads
####
#判断是否转化失败只是一个阈值的假定,这一步可做可不做。
filter_non_conversion --help
filter_non_conversion
-s 单端测序
-p 双端测序
--bam 上一步得到的的bam文件
(snakemake)yulan 10:39:42 ~/wgbs_test/filter
$ cat >filter.sh
filter_non_conversion -p /home/yulan/wgbs_test/duplicate/Testpaired_pe.deduplicated.bam
^C
(snakemake)yulan 10:43:49 ~/wgbs_test/filter
$ nohup bash filter.sh >filter.log &
[1] 3556762
yulan 10:44:25 ~/wgbs_test/filter
$ nohup: ignoring input and redirecting stderr to stdout
- 结果
Testpaired_pe.deduplicated.nonCG_filtered.bam 储存了最后要用于calling的reads; Testpaired_pe.deduplicated.non-conversion_filtering.txt 文件为结果概述; Testpaired_pe.deduplicated.nonCG_removed_seqs.bam储存了转化失败的reads.
(snakemake) yulan 15:22:33 ~/wgbs_test/duplicate
$ ll -h
total 75G
drwxrwxr-x 2 yulan yulan 4.0K 1月 8 15:22 ./
drwxrwxr-x 9 yulan yulan 4.0K 1月 8 10:39 ../
-rw-rw-r-- 1 yulan yulan 13K 1月 8 00:40 duplicate.log
-rw-rw-r-- 1 yulan yulan 185 1月 7 23:49 duplicate.sh
-rw-rw-r-- 1 yulan yulan 3.1K 1月 8 14:29 filter.log
-rw-rw-r-- 1 yulan yulan 108 1月 8 10:43 filter.sh
-rw-rw-r-- 1 yulan yulan 38G 1月 8 00:39 Testpaired_pe.deduplicated.bam
-rw-rw-r-- 1 yulan yulan 38G 1月 8 14:29 Testpaired_pe.deduplicated.nonCG_filtered.bam
-rw-rw-r-- 1 yulan yulan 76M 1月 8 14:29 Testpaired_pe.deduplicated.nonCG_removed_seqs.bam
-rw-rw-r-- 1 yulan yulan 305 1月 8 14:29 Testpaired_pe.deduplicated.non-conversion_filtering.txt
-rw-rw-r-- 1 yulan yulan 307 1月 8 00:39 Testpaired_pe.deduplication_report.txt
(snakemake) yulan 15:20:14 ~/wgbs_test/duplicate
$ cat Testpaired_pe.deduplicated.non-conversion_filtering.txt
Analysed read pairs (paired-end) in file >> /home/yulan/wgbs_test/duplicate/Testpaired_pe.deduplicated.bam << in total: 176706382
Sequences removed because of apparent non-bisulfite conversion (at least 3 non-CG calls in one of the reads): 338999 (0.2%)
filter_non_conversion completed in 0d 3h 45m 24s
3.7 甲基化提取(耗时长)
Bismark 附带一个补充的 bismark_methylation_extractor 脚本,为每个分析的单个 C 提取甲基化。单个 C 的位置将被写入一个新的输出文件,具体取决于其context(CpG、CHG 或 CHH),其中甲基化 Cs 将被标记 (+),非甲基化 Cs 被标记 (-)。 bedGraph 计数输出可用于生成全基因组胞嘧啶报告,该报告显示基因组中每个 CpG(可选每个胞嘧啶)的数量,报告对两条链上的胞嘧啶提供了丰富的信息,因此输出会相当大(约 4600 万个 CpG 位置或>12 亿个人类基因组中的总胞嘧啶位置)。bedGraph 到全基因组胞嘧啶报告的转换也可以使用独立模块 bedGraph2cytosine 单独运行。 双末端读取的另一个有用选项称为“--no_overlap”:指定此选项将仅提取一次双末端读取中间重叠部分的甲基化(使用来自第一个reads的调用,这可能错误率最低)。 默认情况下,bismark_methylation_extractor 区分 CpG、CHG 或 CHH context中的胞嘧啶。如果需要,可以通过指定选项“--merge_non_CpG”将 CHG 和 CHH context合并到一个非 CpG context中(Note:这可能会产生多达几亿行的超大文件)。 有关选项的完整列表,请在命令行输入 bismark_methylation_extractor --help
关键的提取甲基化数据,可以分 2 次进行
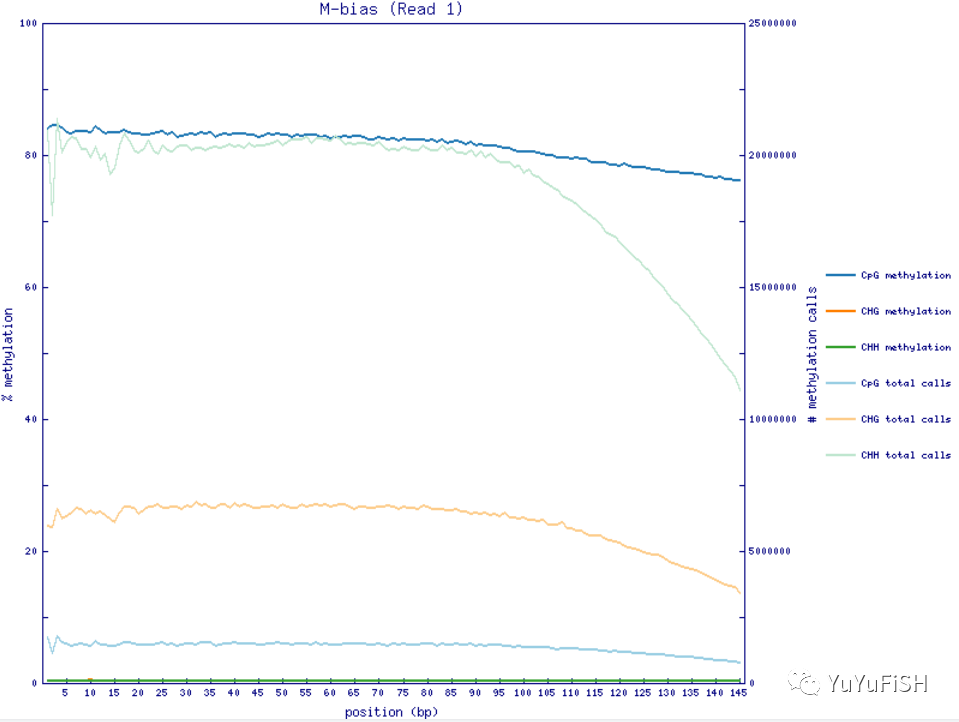
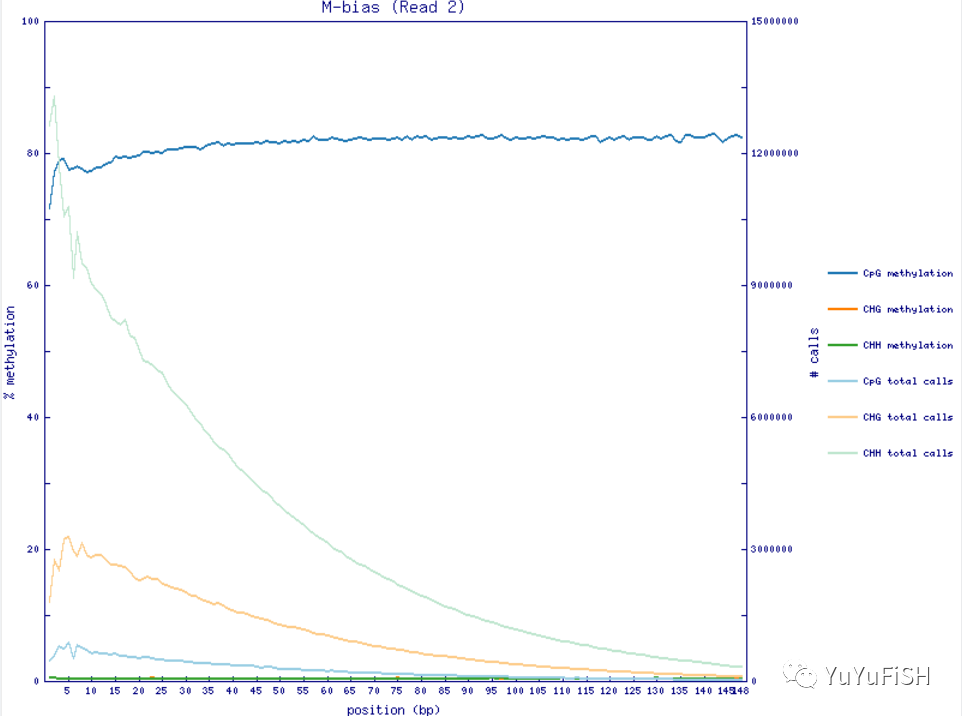
step1.加mbias_only,用生成的结果查看M-bias
# 查看帮助文档
bismark_methylation_extractor --help
# 脚本写进m.sh
#paired
outP=/home/yulan/wgbs_test/extractor
index=/home/yulan/database/hg38
input=/home/yulan/wgbs_test/duplicate/Testpaired_pe.deduplicated.nonCG_filtered.bam
#step1.不加舍弃参数,用生成的结果查看M-bias
bismark_methylation_extractor --comprehensive --report -p --no_overlap --gzip --buffer_size 90G --mbias_only --parallel 30 --output ${outP} --genome_folder ${index} ${input}
# 后台运行
nohup bash m.sh>m.log &
bismark_methylation_extractor
-s 指定单端测序
-p 指定双端测序
--comprehensive 输出CHG CHH CpG的甲基化信息
--no-overlap 去除reads重叠区域的bias,避免因双端读取read_1和read_2理论上的重叠,导致甲基化重复计算。可能会删去相当大部分的数据,对于双端数据的处理,默认情况下此选项处于启用状态,可以使用--include_overlap禁用。
--bedGraph 输出bedGraph文件,其对应的结果文件包含的信息最完整;
--counts 每个C上甲基化reads和非甲基化reads的数目
--buffer_size:甲基化信息进行排序时指定内存,默认为2G
--zero_based:使用基于 0 的基因组坐标而不是基于 1 的坐标,默认值:OFF
--split_by_chromosome:分染色体输出
--report 一个甲基化summay
--cytosine_report 输出全基因组所有CpG
--genome_folder 参考基因组所在目录
input.bam 输入文件
-o <output_dir> 输出路径
-no_header 结果文件不含列名
--ignore_r2 只针对双端测序数据(在第一次提取甲基化水平时不要加上),ignore_r2指的是在提取甲基化 水平时不要提取pair2的前两个位置(类似的参数还有ignore ,ignore_3prime,ignore_3prime_r2。参数选取应M-bias的结果图来确定,详细见下文)--ignore 5:不提取所有pair1的reads 5’端前5个位点,--ignore_r2:不提取所有pair2的reads 5’端前2个位点
--mbias_only (不提取甲基化水平,只给出m-bias信息)
OT – original top strand
CTOT – complementary to original top strand
OB – original bottom srand
CTOB – complementary to original bottom strand
#假如不考虑M-bias直接提取甲基化
/home/yulan/Bismark/bismark_methylation_extractor -p --no_overlap --parallel 30 --bedGraph --buffer_size 90G --cytosine_report --genome_folder /home/yulan/database/hg38
第一次用 --mbias_only不提取数据,只产生 M-bias 质控图
通过Perl模块GD::Graph产生,没有模块的只产生M-bias.txt文件,可用--ignore参数忽略
conda install -c bioconda perl-gdgraph
第二步可利用参数 --ignore和 --ignore_r2分别忽略Reads 5' 端碱基。如果还需要配置 3' 端忽略,用 --ignore_3prim和 --ignore_3prime_r2参数
图中如果看到前几个碱基的甲基化比例非常低,应该是建库末端修复步骤引入的非甲基化 C 碱基,应该移除
step2.依据M-bias,增加参数,提取甲基化
(snakemake) yulan 22:47:34 ~/wgbs_test/extractor
$ cat m.sh
outP=/home/yulan/wgbs_test/extractor
index=/home/yulan/database/hg38
input=/home/yulan/wgbs_test/duplicate/Testpaired_pe.deduplicated.nonCG_filtered.bam
#step2.依据M-bias,增加参数,提取甲基化
bismark_methylation_extractor --comprehensive --report -p --ignore 5 --ignore_r2 2 --no_overlap --bedGraph --cytosine_report --gzip --buffer_size 90G --parallel 30 --output ${outP} --genome_folder ${index} ${input}
结果
(snakemake) yulan 14:34:43 ~/wgbs_test/extractor
$ ls
CHG_context_Testpaired_pe.deduplicated.nonCG_filtered.txt.gz
CHH_context_Testpaired_pe.deduplicated.nonCG_filtered.txt.gz
CpG_context_Testpaired_pe.deduplicated.nonCG_filtered.txt.gz
m.log
m.sh
Testpaired_pe.deduplicated.nonCG_filtered.bedGraph.gz
Testpaired_pe.deduplicated.nonCG_filtered.bismark.cov.gz
Testpaired_pe.deduplicated.nonCG_filtered.CpG_report.txt.gz
Testpaired_pe.deduplicated.nonCG_filtered.cytosine_context_summary.txt
Testpaired_pe.deduplicated.nonCG_filtered.M-bias_R1.png
Testpaired_pe.deduplicated.nonCG_filtered.M-bias_R2.png
Testpaired_pe.deduplicated.nonCG_filtered.M-bias.txt
Testpaired_pe.deduplicated.nonCG_filtered_splitting_report.txt
CHG/CHH/CpG_context_Testpaired_pe.deduplicated.nonCG_filtered.txt.gz
$ less CpG_context_Testpaired_pe.deduplicated.nonCG_filtered.txt.gz | head -6
Bismark methylation extractor version v0.24.0
SRR11243555.3_3/1 + 5 5496733 Z
SRR11243555.12_12/1 + 7 122087247 Z
SRR11243555.5_5/1 + 19 17919675 Z
SRR11243555.5_5/1 + 19 17919662 Z
SRR11243555.5_5/1 + 19 17919630 Z
col1 : 比对上的序列ID
col2 : 基因组正负链:+ -
col3 : 染色体编号
col4 : 染色体位置
col5 : 甲基化C的状态(XxHhZzUu)
#X 代表CHG中甲基化的C
#x 代笔CHG中非甲基化的C
#H 代表CHH中甲基化的C
#h 代表CHH中非甲基化的C
#Z 代表CpG中甲基化的C
#z 代表CpG中非甲基化的C
#U 代表其他情况的甲基化C(CN或者CHN)
#u 代表其他情况的非甲基化C (CN或者CHN)
#CpG:甲基化C下游是个G碱基
#CHH:甲基化C下游的2个碱基都是H(A、C、T)
#CHG:甲基化的C下游的2个碱基是H和G
~bedGraph.gz
$ less Testpaired_pe.deduplicated.nonCG_filtered.bedGraph.gz | head -6
track type=bedGraph
1 10609 10610 0
1 10617 10618 100
1 10620 10621 100
1 10631 10632 100
1 10633 10634 0
#col1 : 染色体编号
#col2 : 胞嘧啶(c)位置信息
#col3 : 胞嘧啶(c)位置信息
#col4 : 甲基化率
~bismark.cov.gz
$ less Testpaired_pe.deduplicated.nonCG_filtered.bismark.cov.gz | head -6
1 10610 10610 0 0 1
1 10618 10618 100 1 0
1 10621 10621 100 1 0
1 10632 10632 100 1 0
1 10634 10634 0 0 1
1 10637 10637 100 1 0
#col1 : 染色体编号
#col2 : 起始位置
#col3 : 终止位置
#col4 : 甲基化率 (col5/col5+col6)
#col5 : 甲基化数目
#col6 : 非甲基化数目
~CpG_report.txt.gz 全基因组胞嘧啶报告
$ less Testpaired_pe.deduplicated.nonCG_filtered.CpG_report.txt.gz | head -6
1 10469 + 0 0 CG CGC
1 10470 - 0 0 CG CGA
1 10471 + 0 0 CG CGG
1 10472 - 0 0 CG CGC
1 10484 + 0 0 CG CGG
1 10485 - 0 0 CG CGG
#col1 : 染色体编号
#col2 : 位置
#col3 : 正负链信息
#col4 : 甲基化碱基数目
#col5 : 非甲基化碱基数目
#col6 : 类型 context (CpG, CHH or CHG)
#col7 : 具体背景 C or CG context (CGX or CHX, X = A, T, C ,G, H = A, T, G)
~cytosine_context_summary.txt
$ less Testpaired_pe.deduplicated.nonCG_filtered.cytosine_context_summary.txt | head -6
upstream C-context full context count methylated count unmethylated percent methylation
A CAA ACAA 2586 1382 65.17
C CAA CCAA 1802 700 72.02
G CAA GCAA 154 158 49.36
T CAA TCAA 10510 3913 72.87
A CAC ACAC 738 400 64.85
CpG、CHG 或 CHH context 中胞嘧啶的甲基化百分比(其中 H 可以是 A、T 或 C)。该百分比是根据以下等式为每个上下文单独计算的:

应该强调的是,甲基化百分比值(context)只是在mapping步骤中直接执行的非常粗略的计算。应用后处理或过滤后的实际甲基化水平可能会有所不同。
~M-bias.txt 每一个甲基化位点的详细信息
$ less Testpaired_pe.deduplicated.nonCG_filtered.M-bias.txt | head -6
CpG context (R1)
================
position count methylated count unmethylated % methylation coverage
1 1433788 271272 84.09 1705060
2 962042 175141 84.60 1137183
3 1497567 272040 84.63 1769607
定义了每一个甲基化位点的详细信息,%methylation就是我们定量常用的beta 值
~_splitting_report.txt 甲基化总体报告
$ cat Testpaired_pe.deduplicated.nonCG_filtered_splitting_report.txt
Testpaired_pe.deduplicated.nonCG_filtered.bam
Parameters used to extract methylation information:
Bismark Extractor Version: v0.24.0
Bismark result file: paired-end (SAM format)
Ignoring first 5 bp of Read 1
Ignoring first 2 bp of Read 2
Output specified: comprehensive
No overlapping methylation calls specified
Processed 176367383 lines in total
Total number of methylation call strings processed: 352734766
Final Cytosine Methylation Report
=================================
Total number of C's analysed: 4516594083
Total methylated C's in CpG context: 190886191
Total methylated C's in CHG context: 3652915
Total methylated C's in CHH context: 11058777
Total C to T conversions in CpG context: 43351315
Total C to T conversions in CHG context: 1037551453
Total C to T conversions in CHH context: 3230093432
C methylated in CpG context: 81.5%
C methylated in CHG context: 0.4%
C methylated in CHH context: 0.3%
M-bias plot