全基因组 - 人类基因组变异分析 (PacBio)(6)-- ANNOVAR
原创全基因组 - 人类基因组变异分析 (PacBio)(6)-- ANNOVAR
原创
如果将个体基因组与参考基因组相比,变异的数量是巨大的。据估计(1),全球范围内人类的基因组中总共有超过8800万个变异(包括约8470万个单核苷酸多态性、360万个短插入/缺失变异和约6万个结构变异)。但如果只考虑你和我两个人,我们基因组上的差别并没有这么多,因为在上述8800万个变异位点上我们的序列很大可能是相同的。实际上,如果我们和人类参考基因组GRch38相比,那么我们的基因组差异大概在400-500万个(其中超过99.9%是单核苷酸多态性和短片段插入缺失变异),手动检查每个位点非常耗时且有些不切实际。
一. 软件介绍
ANNOVAR是由王凯老师编写的一款用于SNP等变异位点注释的软件 (2),在注释软件(Annovar, SnpEff, VEP-Variant Effect Predictor)中相对引用较高。ANNOVAR能够利用最新的数据来分析各种基因组中的遗传变异。 给定一个包含染色体,起点,终点,参考核苷酸与检测核苷酸序列, ANNOVAR可以进行如下的功能注释:
基于基因的注释Gene-based annotation:主要针对SNP或CNV是否引起蛋白编码改变进行注释,可以灵活选用 RefSeq genes, UCSC genes, ENSEMBL genes, GENCODE genes, AceView genes等多种不同来源的基因定义系统。
基于区域的注释Region-based annotation:针对基因组某一特定区域的变异进行注释,例如44个物种的保守区域,预测的转录因子结合位点,GWAS hit, ENCODE H3K4Me1/H3K4Me3/H3K27Ac/CTCF sites,ChIP-Seq peaks, RNA-Seq peaks等。
基于筛选的注释Filter-based annotation:鉴定在特定数据库中记录的变异,例如一个变异是否在dbSNP数据库中有报道,1000基因组计划、NHLBI-ESP 6500外显子或Exome Aggregation Consortium (ExAC)或Genome Aggregation Database (gnomAD)数据库中的等位基因频率,或许多其他特定突变的注释。
给出这个variant的一系列信息,如: population frequency in different populations 和various types of variant-deleteriousness prediction scores, 这些可被用来过滤掉一些公共的及大概 (肯定的成分较大,是most likely) nondeleterious variants. 包括Clinvar, dbSNP, Cosmic, ExAC, gnomAD等等。 鉴定特定数据库中记录的变异,例如,该变异位点是否在dbSNP中有报道,在千人基因组计划中的等位基因频率如何等等 (3)。
二. 软件安装
网站地址:https://annovar.openbioinformatics.org/en/latest/
ANNOVAR是由perl编写的程序,首先通过下载页面(图1),填写注册表格,学术用途可以免费试用,几分钟后邮件会发来下载链接。
将ANNOVAR的压缩包annovar.latest.tar.gz上传至服务器,解压并将路径添加到环境中 (图2)。
$ tar -xzvf annovar.latest.tar.gz
$ echo "PATH=/mnt/data/home/mli/Desktop/Software:$PATH" >> ~/.bashrc && source ~/.bashrc
三. 软件使用
ANNOVAR程序有以下几个模块:
ANNOVAR结构
│ annotate_variation.pl #主程序,功能包括下载数据库,三种不同的注释
│ coding_change.pl #可用来推断突变蛋白质序列
│ convert2annovar.pl #将多种输入格式转为.avinput的程序
│ retrieve_seq_from_fasta.pl #用于自行建立其他物种的转录本
│ table_annovar.pl #一次可完成多种类型的注释
│ variants_reduction.pl #可用来更灵活地定制过滤注释流程
│
├─example #存放示例文件
│
└─humandb #人类注释数据库1. 数据库的下载
对变异进行注释前需要先下载注释数据库:
- 基于基因的注释Gene-based annotation
这里选择ensGene, refGene和knownGene数据库最新更新版本进行下载。
# For gene-based annotation
$ perl annotate_variation.pl --buildver hg38 --downdb ensGene --webfrom annovar humandb/
$ perl annotate_variation.pl --buildver hg38 --downdb refGene --webfrom annovar humandb/
$ perl annotate_variation.pl --buildver hg38 --downdb knownGene --webfrom annovar humandb/--buildver 基因组版本号
--downdb 注释数据库名称
--webfrom 指定数据库来源(ucsc,annovar or URL)
-- 或 - 都行
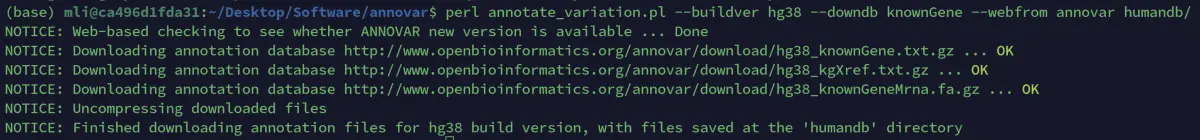
如图3所示,展示了ANNOVAR数据库下载的过程:
- 基于筛选的注释Filter-based annotation
#For Filter-based annotation
$ perl annotate_variation.pl --buildver hg38 --downdb clinvar_20221231 --webfrom annovar humandb/
$ perl annotate_variation.pl --buildver hg38 --downdb avsnp150 --webfrom annovar humandb/2. ANNOVAR使用
ANNOVAR的使用分为annotate_variation.pl单个数据库的注释和table_annovar.pl多个数据库的注释:
# 使用annotate_variation.pl注释单个数据库
$ perl annotate_variation.pl -geneanno -dbtype refGene -out ex1 -builder hg38 example/ex1.avinput humandb/
$ perl annotate_variation.pl -regionanno -dbtype cytoBand -out ex1 -buildver hg38 example/ex1.avinput humandb/
$ perl annotate_variation.pl -filter -dbtype 1000g2012apr_eur -out ex1 -buildver hg38 example/ex1.avinput humandb/
# -geneanno 表示使用基于基因的注释,另有-regionanno表示基于区域的注释,-filter表示基于过滤的注释
# -dbtype 后跟使用的数据库
# -out ex1 表示输出文件以ex1为前缀
# -buildver hg38 表示使用的参考基因组版本为hg38
# humandb/ 放最后,指示数据库的位置
# 使用table_annovar.pl注释多个数据库
$ perl table_annovar.pl example/ex1.avinput humandb/ -buildver hg38 -out final -remove -protocol refGene,cytoBand,clinvar_20200316 -operation g,r,f -nastring NA -csvout
# -buildver hg38 表示使用的参考基因组版本为hg38
# -out final 指定输出文件前缀为final
# -remove 表示删除中间文件
# -protocol 后跟注释来源数据库名称,每个protocal名称或注释类型之间只有一个逗号,并且没有空格
# -operation 后跟指定的注释类型,和protocol指定的数据库顺序是一致的,g代表gene-based、r代表region-based、f代表filter-based
# -nastring NA 表示用NA替代缺省值
# -csvout 表示最后输出.csv文件如果使用单个数据库进行注释annotate_variation.pl, vcf文件需要转换成.avinput。
ANNOVAR使用.avinput格式,最少需要5列,分别代表染色体(Chromosome),起始位置(Start),终止位置(End),参考等位基因(Reference Allele),替代等位基因(Alternative Allele),其他的列作为额外补充信息(可选)。
#vcf to aviput 格式
$ perl ~/Desktop/Software/annovar/convert2annovar.pl \
-format vcf4 -allsample -withfreq -includeinfo \
deepvariant.cohort.vcf.gz \
-outfile deepvariant.cohort.avinput
#对于多样本vcf,-allsample, -withfreq -includeinfo是必须要加的,才能保证样本信息完整3. ANNOVAR实际演示
- 使用annotate_variation.pl注释单个数据库gene-based:
# 使用annotate_variation.pl注释单个数据库,基于基因的注释
$ perl annotate_variation.pl --geneanno --buildver hg38 -dbtype refGene \
~/Desktop/pb_WGS/deepvariant.cohort.avinput --outfile ~/Desktop/pb_WGS/refGene \
humandb/
# -geneanno 表示使用基于基因的注释,另有-regionanno 表示基于区域的注释,-filter表示基于过滤的注释
# -dbtype 后跟使用的数据库
# -out ex1 表示输出文件以ex1为前缀能得到以下文件refGene.variant_function 和 refGene.exonic_variant_function。
refGene.variant_function所有变异的信息 (一共6,982,339个变异),如图4。
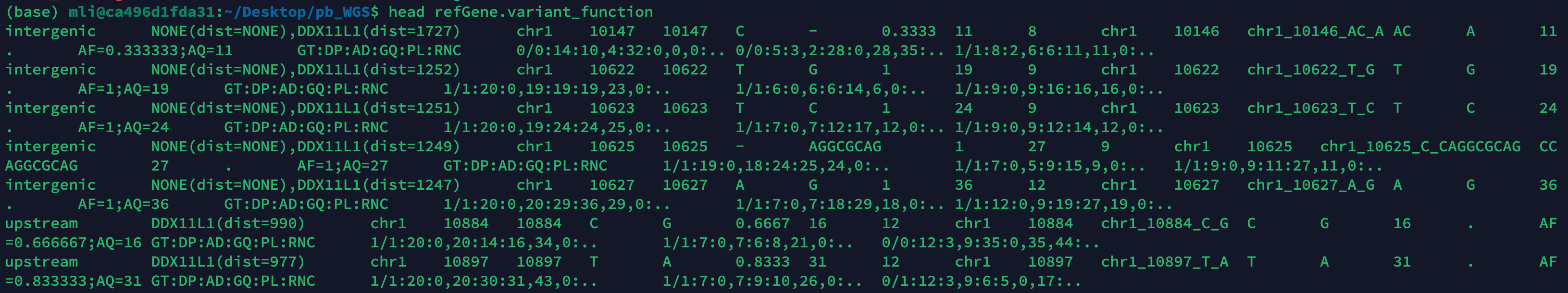
第1列:变异存在位置信息,如intergenic, upstream等。
第2列:基因名,Symbol。
第3列:染色体位置。
第4,5列:突变位置。
第6,7列:参考碱基,突变碱基。
#统计所有变异位置信息
$ cut -f 1 refGene.variant_function | sort | uniq -c
46714 downstream
35227 exonic
44 exonic;splicing
3921596 intergenic
2389507 intronic
27045 ncRNA_exonic
12 ncRNA_exonic;splicing
444673 ncRNA_intronic
137 ncRNA_splicing
2 ncRNA_UTR5
231 splicing
43983 upstream
1657 upstream;downstream
60864 UTR3
10620 UTR5
27 UTR5;UTR3refGene.exonic_variant_function所有外显子变异的信息 (35227+44 = 35271个突变),如图5。
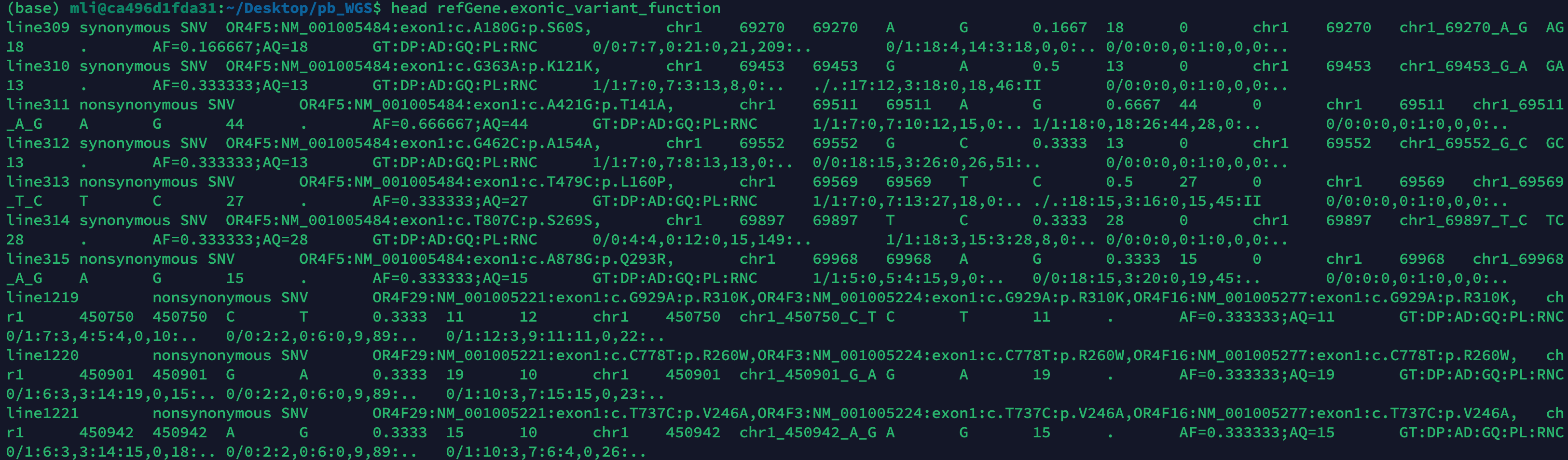
第1列:该变异在input文件的行号。
第2列:对编码基因的影响:frameshift, nonsynonymous,nonframeshift,stopgain,stoploss,unknown。
第3列:突变类型:SNV,insertion, deletion。
第4列:被影响的基因或转录本,其中NM_001005484为refGene编号。
其余列同输入文件。
variant_function注释结果,优先级排序为 exonic = splicing > ncRNA > UTR5/UTR3 > intron > upstream/downstream > intergenic
- 使用annotate_variation.pl注释单个数据库 filter-based:
# 使用annotate_variation.pl注释单个数据库,基于过滤的注释
$ perl annotate_variation.pl --filter --buildver hg38 -dbtype clinvar_20221231 \
~/Desktop/pb_WGS/deepvariant.cohort.avinput --outfile ~/Desktop/pb_WGS/clinvar \
humandb/
# -geneanno 表示使用基于基因的注释,另有-regionanno 表示基于区域的注释,-filter表示基于过滤的注释
# -dbtype 后跟使用的数据库
# -out ex1 表示输出文件以ex1为前缀
#region-based
$ perl annotate_variation.pl -regionanno -dbtype cytoBand -out ex1 -buildver hg19 deepvariant.cohort.avinput humandb/ #region-basedclinvar.hg38_clinvar_20221231_filtered(clinvar中没有报道的位点)和clinvar.hg38_clinvar_20221231_dropped(clinvar中报道的位点,包含其等位基因频率)。
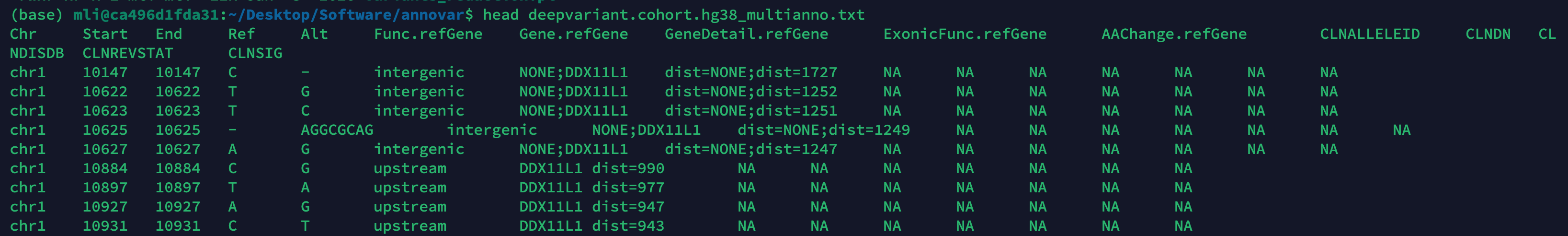
- table_annovar.pl 可以多个数据库同时进行注释:
$ perl table_annovar.pl ~/Desktop/pb_WGS/deepvariant.cohort.avinput humandb/ -buildver hg38 -out deepvariant.cohort -remove -protocol refGene,clinvar_20221231 -operation g,f -nastring NA -polish
#-remove: remove all temporary files
#-operation:g,gene-based; r, region-based; f,filter-based.
#-nastring:没有对应注释,则输出`NA`
#--polish polish the protein notation for indels (such as p.G12Vfs*2)一起注释的结果,如图6:
参考文献:
- 1000 Genomes Project Consortium, 2015. A global reference for human genetic variation. Nature, 526(7571), p.68.
- Wang K, Li M, Hakonarson H.?ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data?Nucleic Acids Research, 2010.
- ANNOVAR | 变异注释【上】-生信师姐
- ANNOVAR | 变异注释【下】-生信师姐
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。