V5版seurat读取不同格式单细胞数据

前情概要
在23年3月份的时候(下意识想说今年了hhh,恍然发现已经24年),菜鸟团作者就整理过不同格式的单细胞数据读取的方法,是基于V4版本的。
当时我在学习单细胞的时候,读取数据都是按照推文里面的方法使用的,也就有了不同格式单细胞数据下载及读取分析流程这篇笔记。
但目前seurat包已经更新到5.0.1版本,更新后使用起来也花了一些时间Seurat包更新与使用初探
虽然感觉在seurat对象结构上,V4和V5版本区别不大——V5和V4版Seurat对象内部结构对比详细版,但是在读取数据的时候,V4和V5的区别还是有点明显的。
如果是单个样品,直接读取进来然后创建seurat对象即可:初试Seurat的V5版本
主要区别在于,V4版本中一般是循环读取样品,使用CreateSeuratObject创建seurat对象,然后使用merge对数据进行整理。
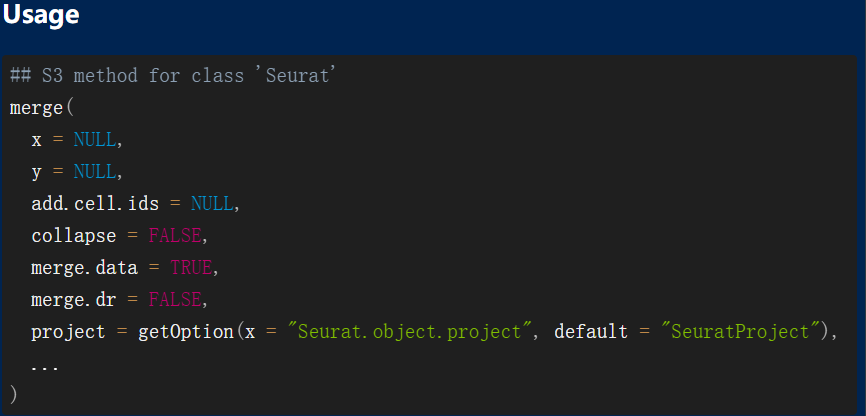
而在V5版的seurat中如果是分开读取多个文件后,再使用merge函数其实并没有把每个样品的表达量矩阵merge。那我们可以先把多个样品合并成为了一个超级大的表达量矩阵,并使其行名为基因名,列名为barcodes信息,后面直接针对它来使用CreateSeuratObject函数去构建Seurat对象,就是完美的下游分析的输入数据啦。
使用Seurat的v5来读取多个不是10x标准文件的单细胞项目
不同格式单细胞多数据读取方法
读取数据进行分析之前,我们需要安装加载需要的R包,之前的推文也整理过需要安装的系列R包
library(COSG)
library(harmony)
library(ggsci)
library(dplyr)
library(future)
library(Seurat)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(ggplot2)
library(patchwork)
library(stringr)
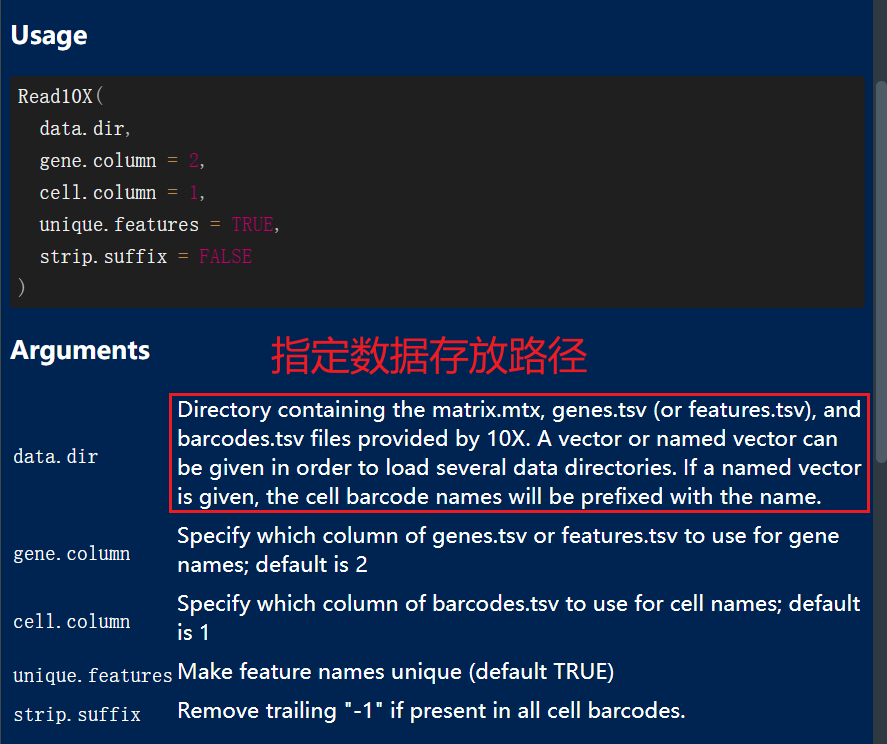
10X标准格式
如果是10X标准格式的多个数据,那我们使用Read10X()函数将多个数据读取进来,再创建seurat对象即可
##10X标准格式
#单个样品的数据V4和V5读取进来没有太大差异
#置顶
samples=list.files("./GSE212975/")
samples
dir <- file.path('./GSE212975/',samples)
names(dir) <- samples
#读取数据创建Seurat对象
counts <- Read10X(data.dir = dir)
sce.all = CreateSeuratObject(counts,
min.cells = 5,
min.features = 300 )
dim(sce.all) #查看基因数和细胞总数
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
table(sce.all@meta.data$orig.ident) #查看每个样本的细胞数
head(sce.all@meta.data)
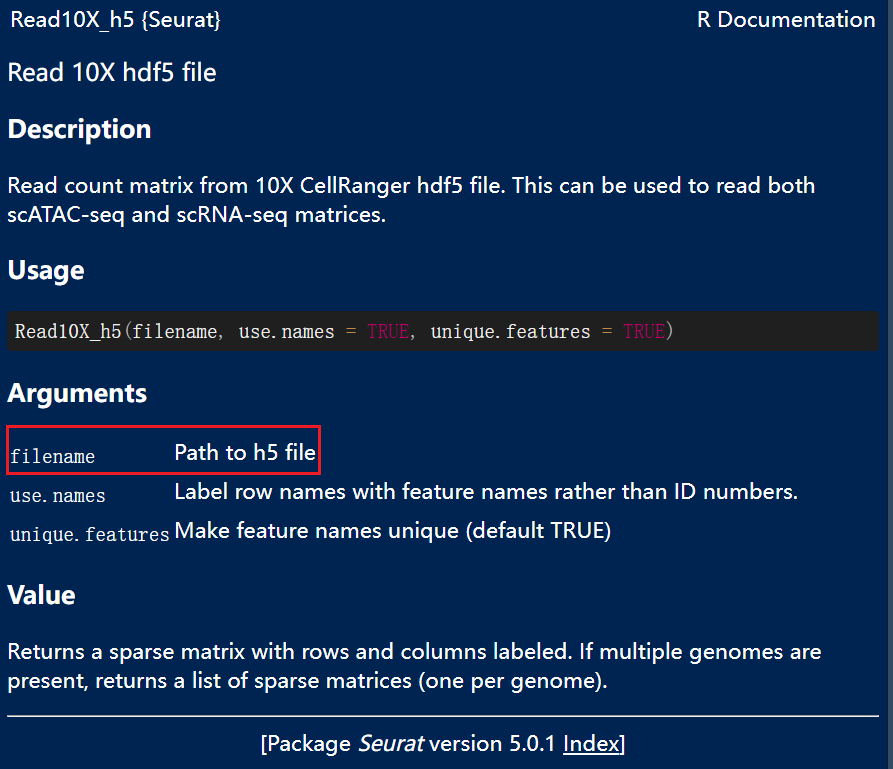
h5格式
h5格式其实也有对应的函数Read10X_h5()可以直接读取,但是Read10X_h5使用循环读取多个数据文件,会返回一个list,需要手动整合一下
#加载需要的R包
library(hdf5r)
library(stringr)
library(data.table)
#设置文件路径
dir='./GSE159115_RAW/'
samples=list.files( dir )
samples
#读取h5格式文件
sceList = lapply(samples,function(pro){
print(pro)
counts = Read10X_h5( file.path(dir,pro))
return(counts)
})
#更改列名,让barcodes具有特异性
samples<-str_split(samples,"_",simplify = T)[,1]
for (i in 1:length(sceList)) {
col<-colnames(sceList[[i]])
colnames(sceList[[i]])<-paste0(samples[i],"_",col)
}
#数据整合后创建seurat对象
merge <- do.call(cbind,sceList)
sce =CreateSeuratObject(counts = merge ,
min.cells = 5,
min.features = 300 )
#查看seurat对象结构
sce
as.data.frame(sce@assays$RNA$counts[1:10, 1:2])
head(sce@meta.data, 10)
table(sce@meta.data$orig.ident)
txt.gz以及csv.gz格式
txt.gz以及csv.gz格式读取方法差不太多,所以就放到一类下面啦。
参考推文:使用Seurat的v5来读取多个不是10x标准文件的单细胞项目
txt.gz格式
dir='./GSE167297/'
samples=list.files( dir ,pattern = 'gz')
samples
library(data.table)
#先把矩阵读取进来
ctList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_CountMatrix.txt.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
return(ct)
})
#将数据整合为一个大list
lapply(ctList, dim)
tmp =table(unlist(lapply(ctList, rownames)))
cg = names(tmp)[tmp==length(samples)]
bigct = do.call(cbind,
lapply(ctList,function(ct){
ct = ct[cg,]
return(ct)
}))
dim(bigct)
#创建seurat对象
sce.all=CreateSeuratObject(counts = bigct,
min.cells = 5,
min.features = 300)
sce.all
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
csv.gz格式
library(data.table)
dir='./GSE129516_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread( file.path(dir,pro),data.table = F)
ct[1:4,1:4]
ct[nrow(ct),1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_filtered_gene_bc_matrices.csv.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
ct[1:4,1:4]
return(ct)
})
lapply(sceList, dim)
tmp =table(unlist(lapply(sceList, rownames)))
cg = names(tmp)[tmp==length(samples)]
bigct = do.call(cbind,
lapply(sceList,function(ct){
ct = ct[cg,]
return(ct)
}))
dim(bigct)
#创建seurat对象
sce.all=CreateSeuratObject(counts = sceList,
min.cells = 5,
min.features = 300)
sce.all
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
综上就是目前学会的不同格式单细胞多数据读取方法啦!
一个10X小特例
一般而言10X的标准格式会按照样品名对应三个文件:barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,但GSE184708有点小例外,虽然有二十个samples,但是数据是整合的
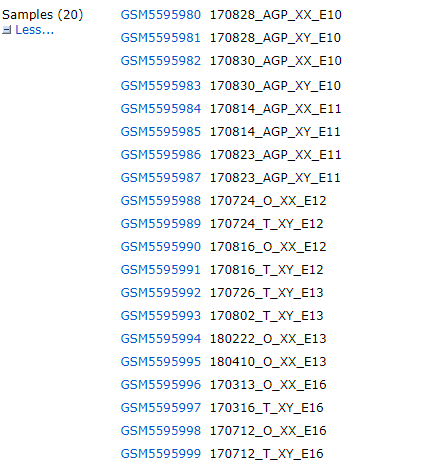
下载数据之后,分别读取barcodes、genes以及matrix矩阵文件,将三个文件对应整理成一个规范的带有行列名的矩阵,再创建seurat对象即可
#加载需要的R包
library(data.table)
library(Matrix)
#将三个文件按照对应的格式分别读取进来
mtx=readMM( "./GSE184708/GSE184708_raw_counts_gonad_all_samples_XX_XY_E10_to_E16.mtx.gz" )
mtx[1:4,1:4]
dim(mtx)
cl=fread( "./GSE184708/GSE184708_mayere_barcodes.tsv.gz" ,
header = F,data.table = F )
head(cl)
rl=fread( "./GSE184708/GSE184708_mayere_genes.tsv.gz" ,
header = F,data.table = F )
head(rl)
#整合矩阵信息
colnames(mtx)=cl$V1
rownames(mtx)=rl$V1
#创建seurat对象
sce.all=CreateSeuratObject(counts = mtx ,
project = 'mouse',
min.cells = 5,
min.features = 300 )
#分组
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
library(stringr)
phe=str_split(rownames(sce.all@meta.data),'_',simplify = T)
head(phe)
table(phe[,2])
sce.all$group=phe[,2]
table(phe[,3])
sce.all$sex=phe[,3]
table(phe[,1])