10X空间转录组数据中研究并可视化体细胞突变
原创10X空间转录组数据中研究并可视化体细胞突变
原创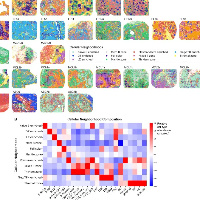
追风少年i
发布于 2024-03-26 18:01:25
发布于 2024-03-26 18:01:25
作者,Evil Genius
关于单细胞数据call snp的分析已经分享了很多了,文章也发了nature,如下:
关于空间转录组和SNP的一些答疑
单细胞、空间、外显子解析TP53突变重构肺腺癌细胞图谱
多组学(单细胞、空间转录+蛋白、外显子、甲基化)揭示神经母细胞瘤异质性图谱
单细胞、空间、外显子分析方法更新
单细胞、空间、外显子多组学分析探讨
单细胞、空间、外显子多组学分析揭示了早期肺腺癌(LUAD)发展过程中上皮细胞的状态和转录组特征
外显子call突变的流程我也分享过了,注意这是流程,跟脚本不是一回事
外显子(WES)panel分析基础篇
cfDNA(带UMI标签的fq数据--WES)的生信处理call突变流程
肿瘤突变负荷(TMB)及计算方法
OncoKB数据库介绍及爬虫爬取位点用药信息
外显子(wes)panel数据分析OncoKB注释
外显子(panel)数据分析的初步解读
NGS基因测序(panel)报告解读数据库汇总
那么接下来,我们要分享的内容,就是空间数据和SNP的关系。如果可以看到突变位点的空间信息,那就是一个发表nature的方向。
人体是由遗传上不同的细胞拼接而成的,这是细胞一生中不断积累体细胞变化的结果。大多数突变是中性的,不影响细胞表型。然而,一些突变降低了细胞的适应性,促进了衰老的过程,而另一些突变增加了细胞的适应性,这最终会导致癌症。解决突变细胞在病变和正常组织中的空间分布可以揭示肿瘤进化的早期阶段。体细胞突变标志着维持正常组织病理表型的部分转化细胞的克隆群体。此外,晚期肿瘤通常由基因上不同的亚克隆组成。确定这些亚克隆的空间分布有助于确定影响基因表达异质性的遗传和非遗传因素的相对贡献。这里,研究在Visium平台生成的空间转录组数据中可视化体细胞突变。
对于单细胞RNA测序数据,已经开发了几种工具来检测体细胞点突变.
DNA水平的拷贝数变化也可以从RNA测序数据推断出来。人们开发了一个名为intercnv的程序,用于从单细胞RNA测序数据中获得拷贝数改变,最近该程序被应用于空间转录组数据集。另一个程序,称为STARCH,也可以从空间转录组数据推断拷贝数信息。这两个软件包都计算基因表达在转录组中的移动平均值,以产生拷贝数估计
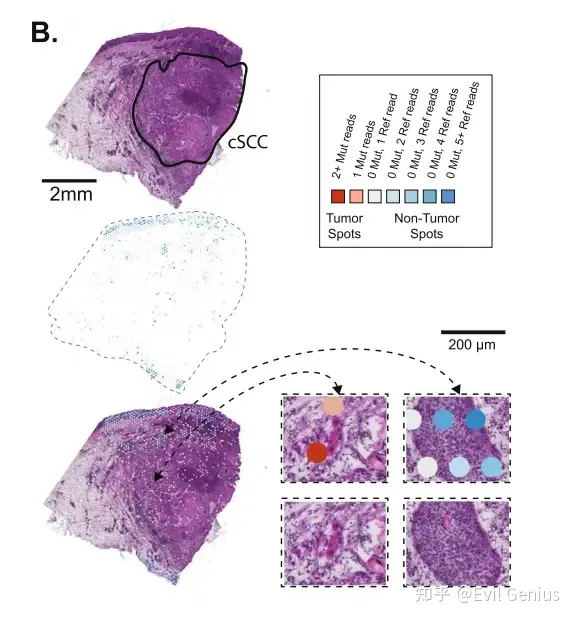
Visualization of point mutations in fresh?frozen Visium data
新鲜冷冻的Visium平台捕获并测序来自polyA尾部的转录本,将突变检测限制在表达基因的3 '端附近。
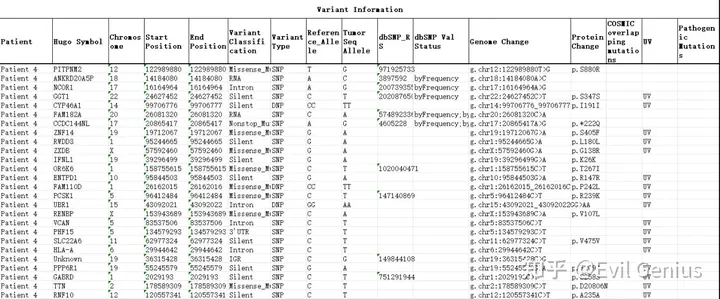
接下来,绘制肿瘤组织中这些突变位点的测序reads,一个spot有一个或多个突变位点来表征肿瘤细胞,如果一个SPOT有5个或更多的参考位点而没有任何突变等位基因,认为它不太可能含有肿瘤细胞。称为无肿瘤点的较高阈值反映了从杂合突变中取样野生型等位基因的可能性。含有突变reads的SPOT大多定位于组织病理学上标记为肿瘤的区域。
在正常组织中观察到少量突变位点。
在组织学上良性组织中检测到的突变reads可能起源于邻近肿瘤组织中的RNA分子,并在杂交过程中扩散到其他spot。为了模拟扩散的程度,检查了没有被任何组织覆盖的斑点的总读取计数。被组织覆盖的spot的中位数读数为16,709,而组织外的spot的中位数读数只有213。在未被组织覆盖的spot上存在测序reads表明mRNA或条形码确实发生了一定程度的扩散,但mRNA丰度比组织spot高近两个数量级。接下来,检查了组织外区域的突变reads计数,发现了带有突变的痕量读取。非癌组织区域的突变reads密度(每平方毫米1.45个突变点)比组织区域外的突变reads密度高约10倍。综上所述,mRNA的扩散不太可能解释正常区域突变reads的数量。
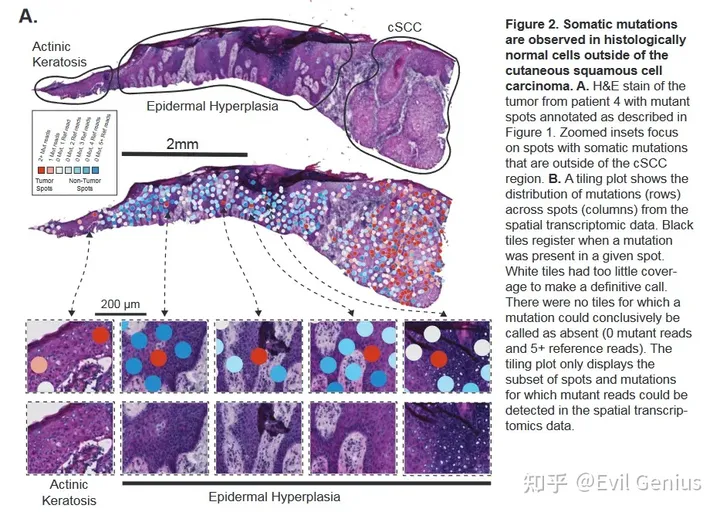
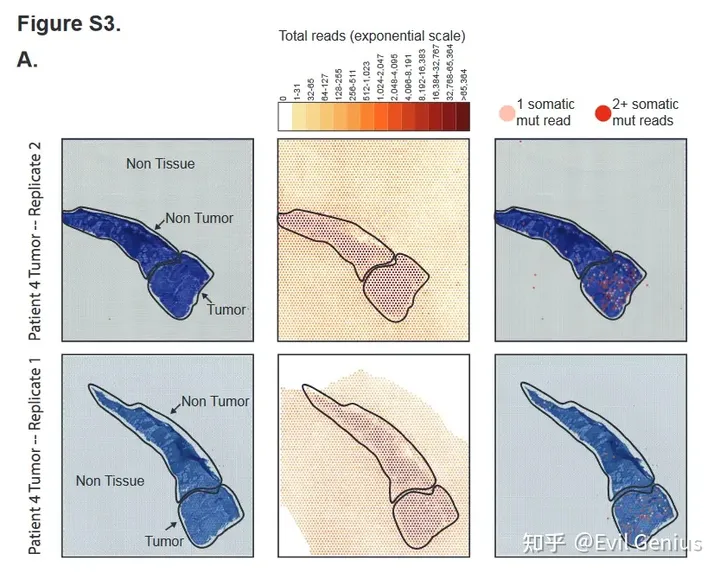
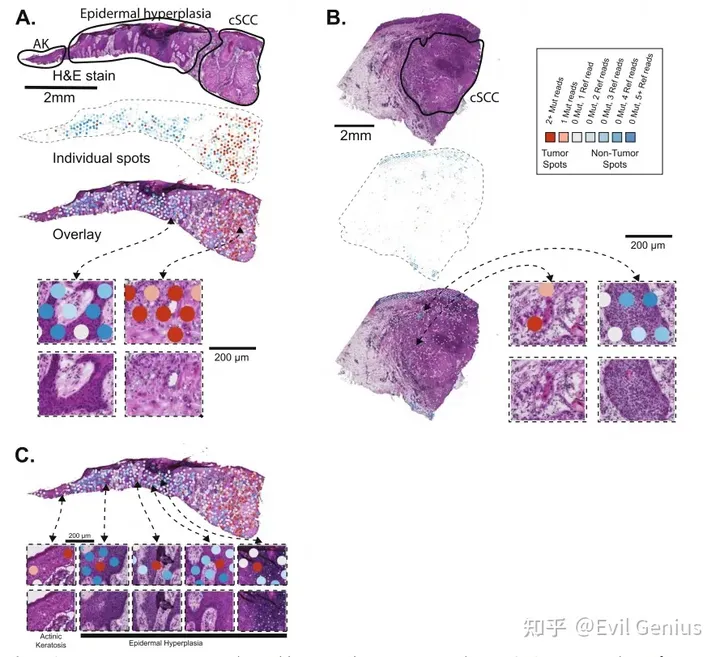
Somatic point mutations are detectable in spatial transcriptomics data
Visualization of copy number alterations in 10X Genomics Visium data
虽然基因表达水平受到许多变量的影响,但通过在染色体滑动窗口中平均多个相邻基因的转录本水平,可以从RNA测序数据中推断出潜在基因的DNA拷贝数。这种策略减少了个体基因表达的可变性,从而揭示了基因表达的变化,在更大的基因组片段中,这通常伴随着拷贝数的改变。这种方法为CNVkit-RNA,当转录本的基因表达与癌症基因组图谱项目中潜在基因的拷贝数变化高度相关时,它赋予转录本更多的拷贝数调用权重。
使用CNVkit-RNA从单个SPOT推断拷贝数信息。将CNVkit-RNA与intercnv和STARCH进行比较。
FFPE样本进行检测CNV
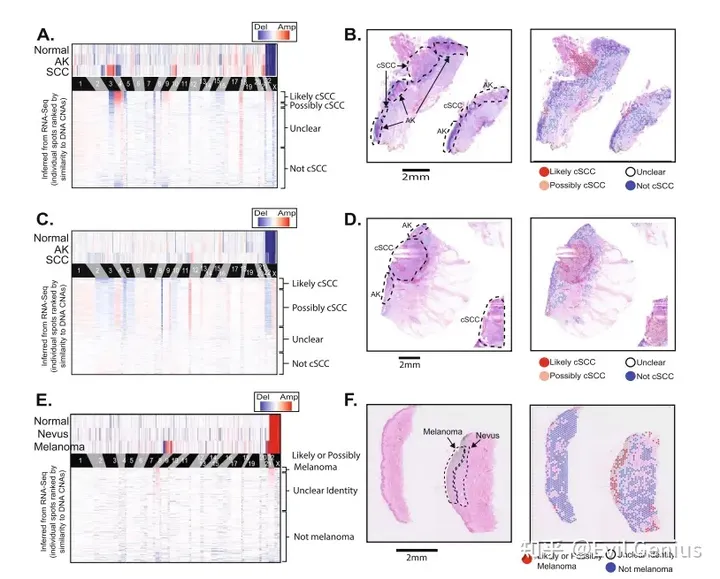
Copy number alterations are detectable in tumors profiled on the FFPE-Visium platform.
10X Genomics Visium数据中等位基因失衡的可视化
接下来测试了等位基因失衡是否可以在空间转录组数据中检测到。从患者正常组织的体细胞DNA测序数据中鉴定出杂合snp。还计算了肿瘤DNA测序数据中与每个等位基因对应的read数,并将数量较多的等位基因指定为“主要”等位基因。绘制了每个SNP和每个spot的主要等位基因和次要等位基因的reads映射比例。如果一个SNP显示单等位基因表达,那么所有的读取将映射到主要或次要等位基因,在散点图中明显具有1:0或0:1的读取比例。单等位基因表达在低表达基因中最常见,正如预期的那样,当采样低读数时,变异性更高。
作为基准,测量了患者的x染色体上杂合snp的等位基因表达,该患者为女性。分析观察到x染色体snp的单等位基因表达,与预期的由一条x染色体失活导致的沉默模式一致。x染色体失活在发育过程中随机发生,导致组织中的马赛克沉默模式。虽然一个SPOT可能覆盖两个不同x染色体失活的细胞群体,但先前的研究表明,共享x染色体失活的细胞的典型克隆大小比Visium阵列的spot大小要大得多,这主要是由于x染色体失活发生在发育的早期阶段。与此一致的是,邻近的spot也倾向于表达相同的等位基因,这支持了马赛克克隆在成年组织中占据相当大体积的观点。XG和RPS4X基因的x染色体snp是异常值,因为它们保留了双等位基因的表达,但这是意料之中的,因为已知这些基因可以逃避x染色体失活。
接下来,在患者的肿瘤spot中测量了3q染色体上杂合snp的空间转录组学数据中的等位基因不平衡。DNA测序数据检测到该区域的等位基因不平衡,可能是由潜在的拷贝数增加引起的。与这一观察结果一致的是,相应的主要等位基因在这些snp的肿瘤细胞上的spot上优先表达。其他染色体区域的等位基因不平衡差异较小,无法可靠地检测到这两种肿瘤。
最后,以无偏的方式探讨了等位基因的不平衡。免疫球蛋白重链位点上存在多个高表达杂合snp,这些snp只在一个等位基因上表达。免疫球蛋白基因在B细胞成熟过程中经历体细胞重排,重排后,未重排的等位基因沉默(这一观察结果被称为“等位基因排斥”)。等位基因排斥确保成熟B细胞产生单一抗体。高水平单等位基因表达的spot定位于肿瘤周围,在免疫细胞密度增加的区域。需要免疫球蛋白重链和轻链mRNA的全长序列来组装VDJ重排,并精确描述B细胞不同区域之间的克隆关系。然而,这里观察到的等位基因排除表明,B细胞的克隆群体包围了肿瘤。
结论
建立了三种类型的遗传改变——体细胞突变、体细胞拷贝数改变和种系多态性——可以在空间转录组学数据中检测到。其中,体细胞点突变在标记具有潜在改变的细胞方面提供了高特异性。然而,检测点突变需要匹配DNA测序数据,这在基于探针的平台(如FFPE-Visium)上是不可能的。此外,点突变的检测并不敏感,因为需要对突变碱基对进行足够的覆盖。长读测序技术可以通过覆盖每个基因的全长而不是3 '端来提高空间转录组数据中突变检测的灵敏度。
代码示例在STmut
参考文章在STmut: a framework for visualizing somatic alterations in spatial transcriptomics data of cancer
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录