生信技能树 Day8 9 GEO数据挖掘 基因芯片数据
原创生信技能树 Day8 9 GEO数据挖掘 基因芯片数据
原创
生信技能树
图表介绍
- 热图
- 散点图
- 箱线图
- 火山图
- 理解logFC
- 主成分分析 PCA样本聚类图
基因芯片差异分析的起点是一个取过log的表达矩阵,得到数据后先看下有没有取log
GEO背景知识
数据库介绍
分析思路
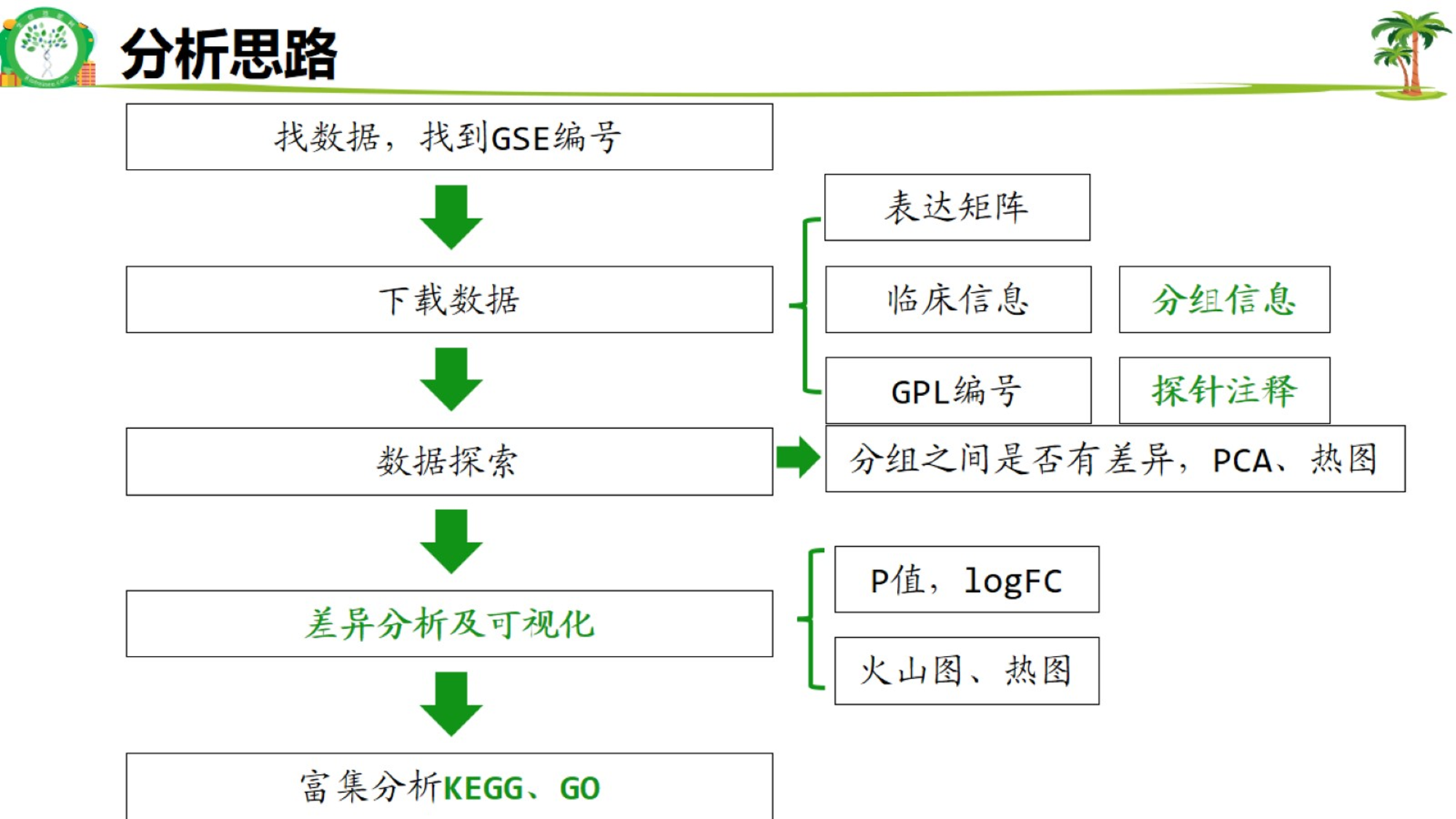
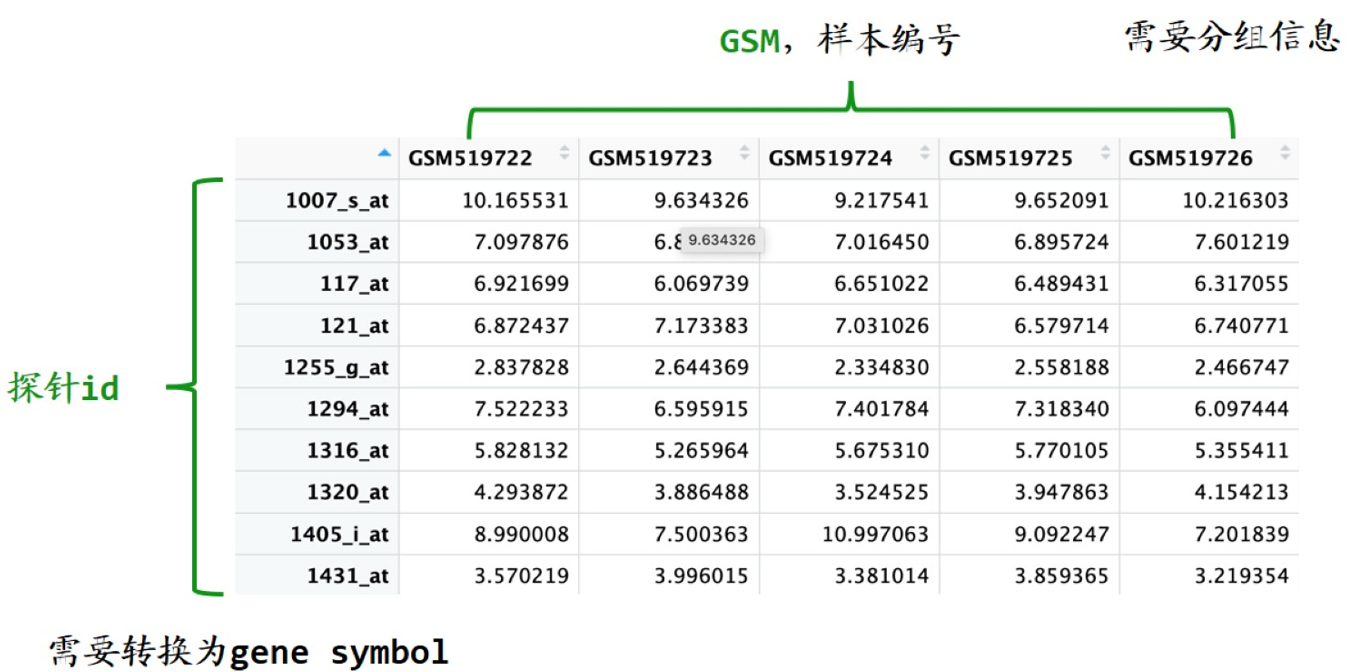
表达矩阵
代码分析流程
数据要求
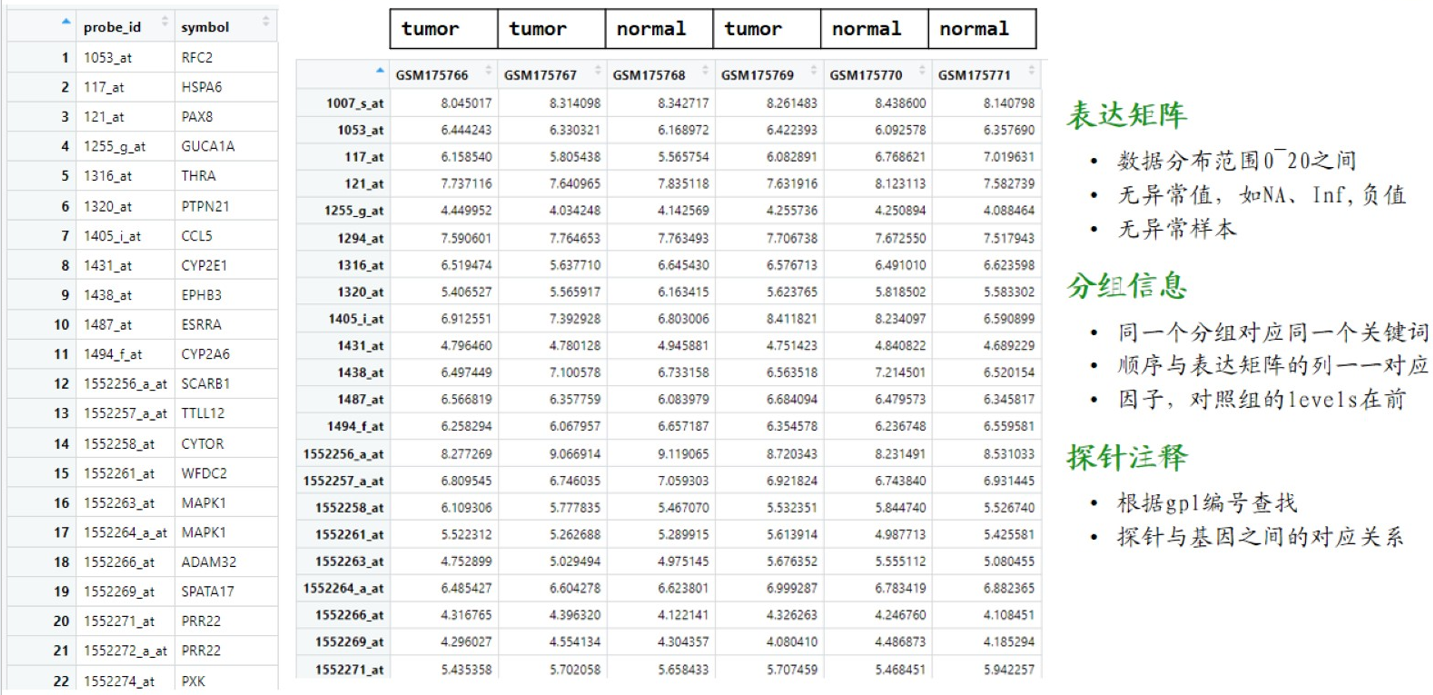

分组信息和探针注释重点学习
安装包
options("repos"="https://mirrors.ustc.edu.cn/CRAN/")
if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
cran_packages <- c('tidyr',
'tibble',
'dplyr',
'stringr',
'ggplot2',
'ggpubr',
'factoextra',
'FactoMineR',
'devtools',
'cowplot',
'patchwork',
'basetheme',
'paletteer',
'AnnoProbe',
'ggthemes',
'VennDiagram',
'tinyarray')
Biocductor_packages <- c('GEOquery',
'hgu133plus2.db',
'ggnewscale',
"limma",
"impute",
"GSEABase",
"GSVA",
"clusterProfiler",
"org.Hs.eg.db",
"preprocessCore",
"enrichplot")
for (pkg in cran_packages){
if (! require(pkg,character.only=T,quietly = T) ) {
install.packages(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
for (pkg in Biocductor_packages){
if (! require(pkg,character.only=T,quietly = T) ) {
BiocManager::install(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
#前面的所有提示和报错都先不要管。主要看这里
for (pkg in c(Biocductor_packages,cran_packages)){
require(pkg,character.only=T)
}
#没有任何提示就是成功了,如果有warning xx包不存在,用library检查一下。
#library报错,就单独安装。查找和下载数据
以GSE7305为例
网站点击链接下载
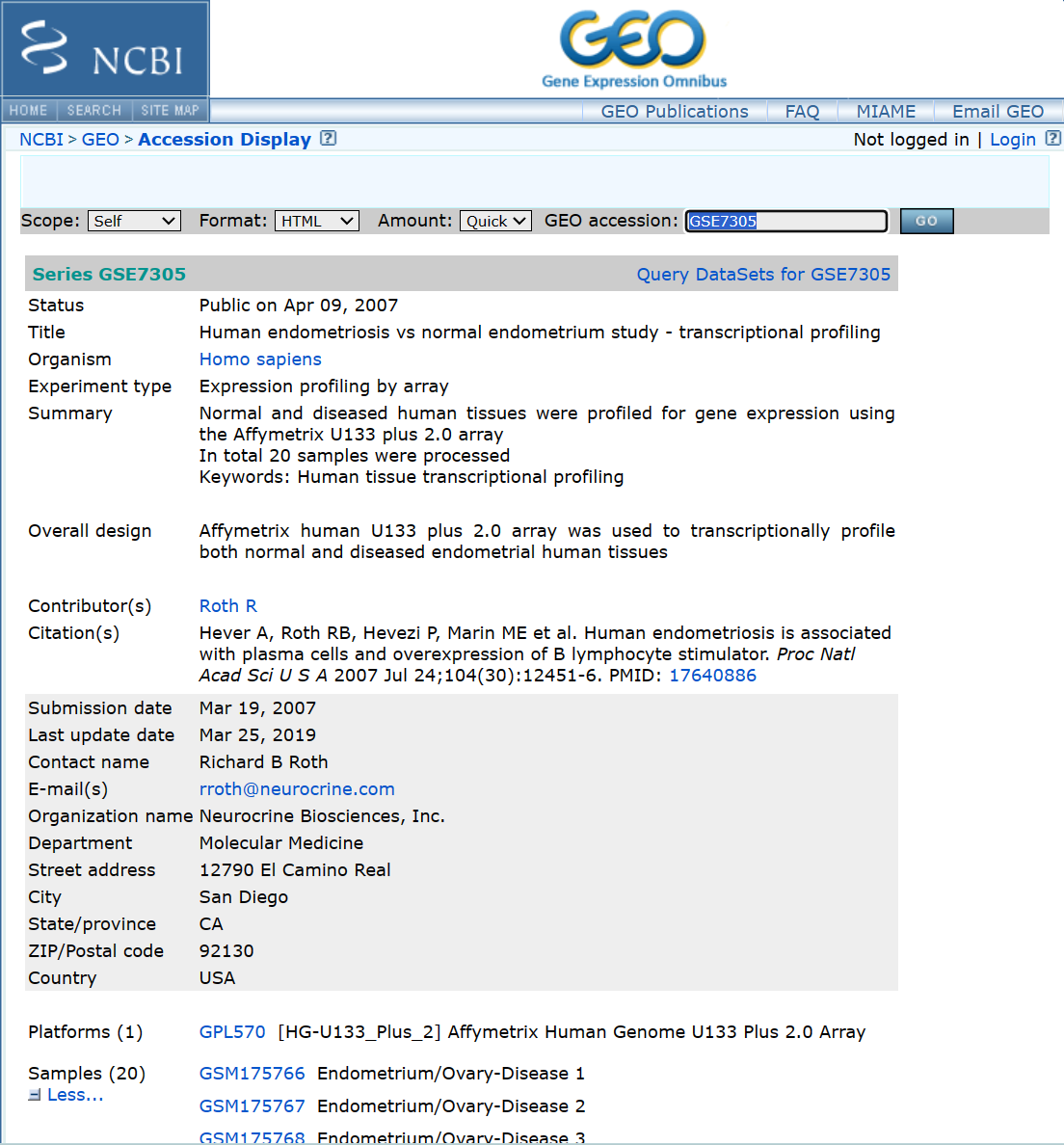

代码下载
#打破下载时间的限制,改前60秒,改后10w秒
options(timeout = 100000)
options(scipen = 20)#不要以科学计数法表示
#传统下载方式
library(GEOquery)
eSet = getGEO("GSE7305", destdir = '.', getGPL = F)
#网速太慢,下不下来怎么办
#1.从网页上下载/发链接让别人帮忙下,放在工作目录里
#2.试试geoChina,只能下载2019年前的表达芯片数据
#library(AnnoProbe)
#eSet = geoChina("GSE7305") #选择性代替第7行什么是eSet
#研究一下这个eSet
class(eSet)
length(eSet)
eSet = eSet[[1]]
class(eSet)
一种R对象,annotation探针注释编号
有时eSet里面有两个对象,可以到网页看一下,可能是因为测了两种芯片,我们分开分析就好。
(1)提取表达矩阵exp
exp <- exprs(eSet) # exprs 提取数据的函数
dim(exp) # 多少行多少列
range(exp) # 看数据范围决定是否需要log,是否有负值,异常值 log后一般是0-13
exp = log2(exp+1) #需要log才log 约定俗成的+1
boxplot(exp,las = 2) # 看是否有异常样本 样本太多只画前20个就可以> #(1)提取表达矩阵exp
> exp <- exprs(eSet)
> dim(exp)
[1] 54675 20
> range(exp)#看数据范围决定是否需要log,是否有负值,异常值
[1] 5.020951 22011.934000
> exp = log2(exp+1) #需要log才log
> boxplot(exp,las = 2) #看是否有异常样本
> 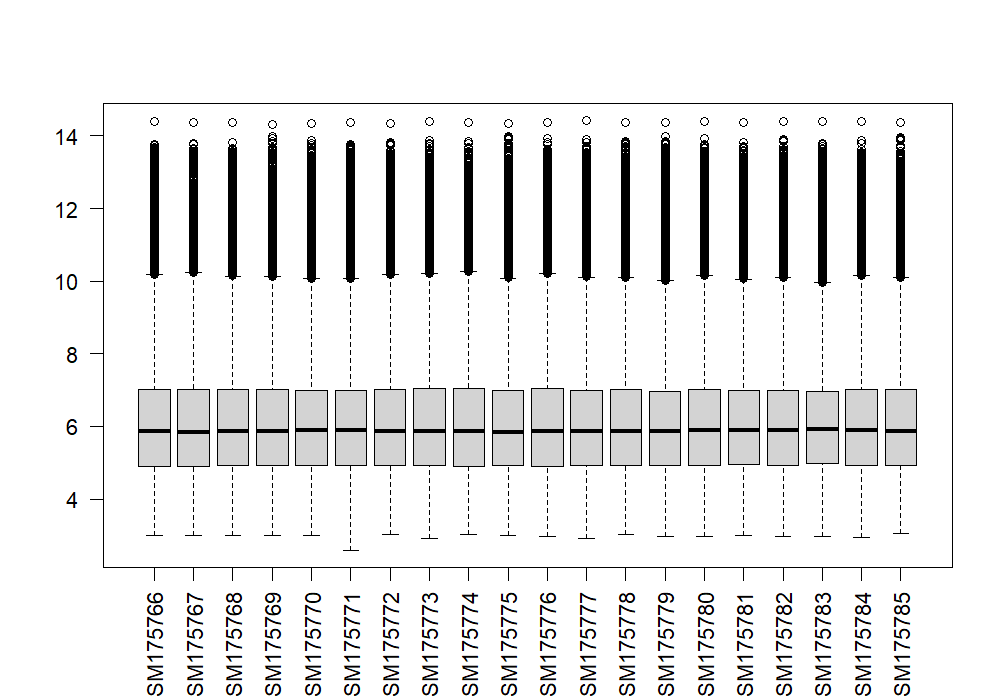
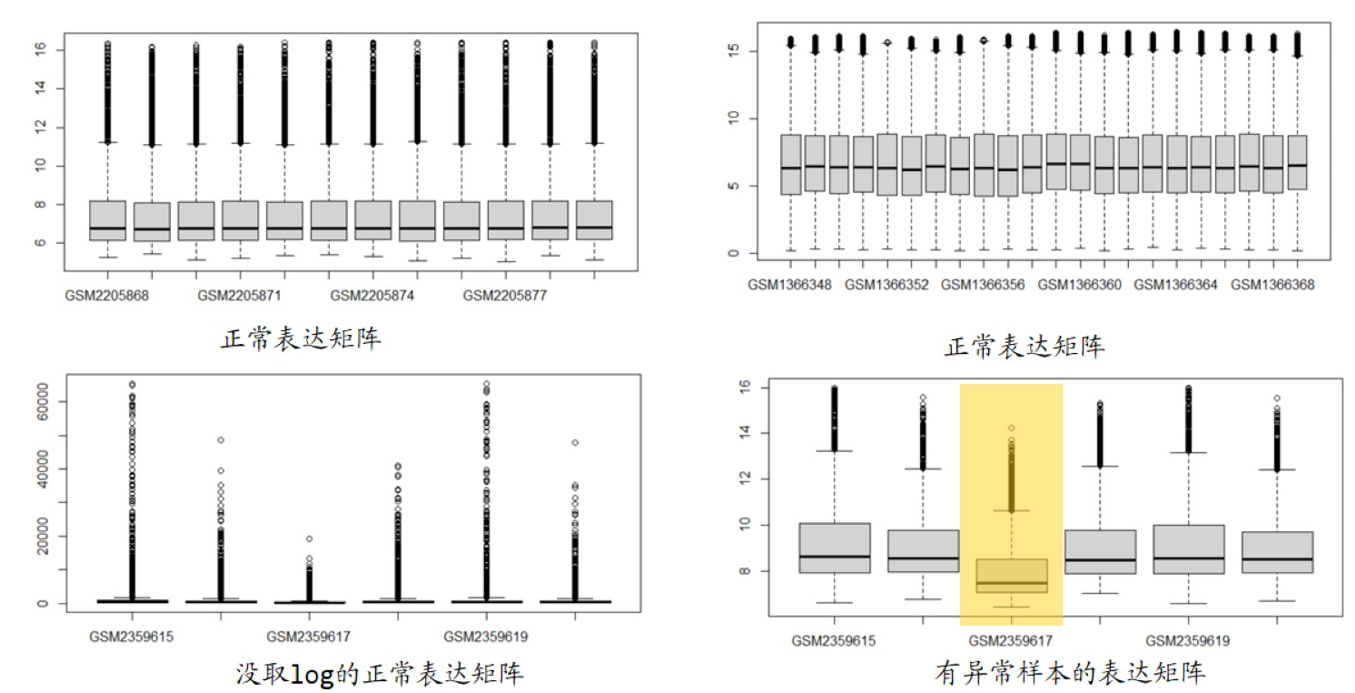
处理异常样本的方法?

关于表达矩阵里的负值

(2)提取临床信息
pd <- pData(eSet) # 找分组信息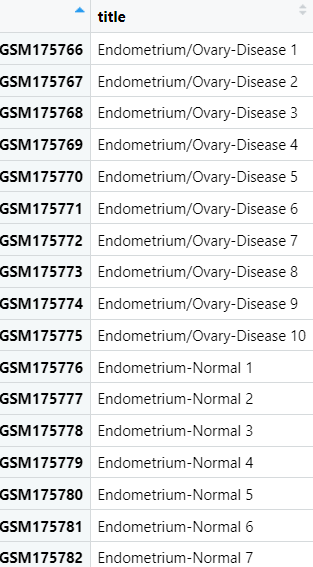
(3)让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) {
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}有多个分组,怎么提取两个分组
#现编一个三分组
pd$group = rep(c("group1","group2","group3"),times = c(6,6,8))
#假如需要从多个分组里面取两个分组对应的行
library(stringr)
k = str_detect(pd$group,"group1|group2");table(k)
pd = pd[k,](4)提取芯片平台编号,后面要根据它来找探针注释
gpl_number <- eSet@annotation;gpl_number
save(pd,exp,gpl_number,file = "step1output.Rdata") # 保存变量,后续进行下一步原始数据处理的代码,按需学习
https://mp.weixin.qq.com/s/0g8XkhXM3PndtPd-BUiVgw
Group(实验分组)和ids(探针注释)
rm(list = ls())
load(file = "step1output.Rdata")1.Group----------------------------------------------------------------------
三种分组方法
library(stringr)
# 标准流程代码是二分组,多分组数据的分析后面另讲
# 生成Group向量的三种常规方法,三选一,选谁就把第几个逻辑值写成T,另外两个为F。如果三种办法都不适用,可以继续往后写else if
if(F){
# 第一种方法,有现成的可以用来分组的列
Group = pd$ #列名
}else if(F){
# 第二种方法,眼睛数,自己生成
Group = rep(c("Disease","Normal"),each = 10)
# rep函数的其他用法?相间、两组的数量不同?
}else if(T){
# 第三种方法,使用字符串处理的函数获取分组
k = str_detect(pd$title,"Normal");table(k)
Group = ifelse(k,"Normal","Disease")
}
data.frame(pd$title,Group)# 检查分组对不对

转换为因子
# 需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = factor(Group,levels = c("Normal","Disease"))
Group> Group
[1] Disease Disease Disease Disease Disease Disease Disease Disease Disease Disease Normal Normal
[13] Normal Normal Normal Normal Normal Normal Normal Normal
Levels: Normal Disease
插播:转换为因子函数levels
# R program to set levels of a factor
??
# Creating a factor
gender <- factor(c("female", "male", "male", "female"));?
gender
??
# Calling levels() function
# to set the levels
levels(gender) <- c("abc", "def")
gender[1] female male male female
Levels: female male
[1] abc def def abc
Levels: abc def2.探针注释的获取---------------------------------------------------------------------------
什么是探针注释:就是探针和基因的对应关系,随着研究的发展会不断更新,所以要查最新的注释。注释来源有4种:Bioconductor注释包,GPL页面表格文件解析,官网下载对应产品注释表格,自主注释
#捷径
library(tinyarray)
find_anno(gpl_number) #辅助写出找注释的代码这里可能返回三种情况
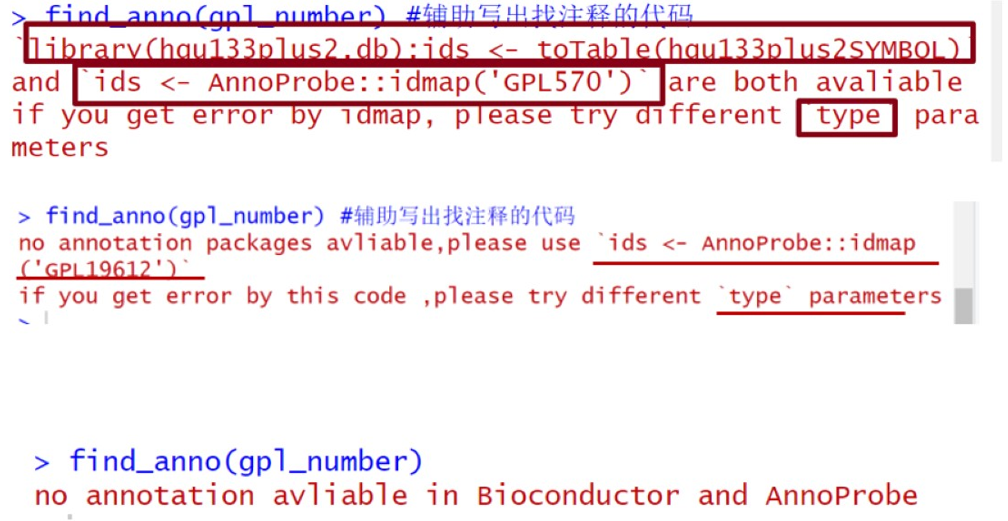
第一、二种情况,按返回的提示复制框中代码运行
若报错显示library的包不存在就自己装一下
library(hgu133plus2.db);ids <- toTable(hgu133plus2SYMBOL) 或
ids <- AnnoProbe::idmap('GPL570')如果使用复制下来的AnnoProbe::idmap('xxx')代码发现报错了,请注意尝试不同的type参数
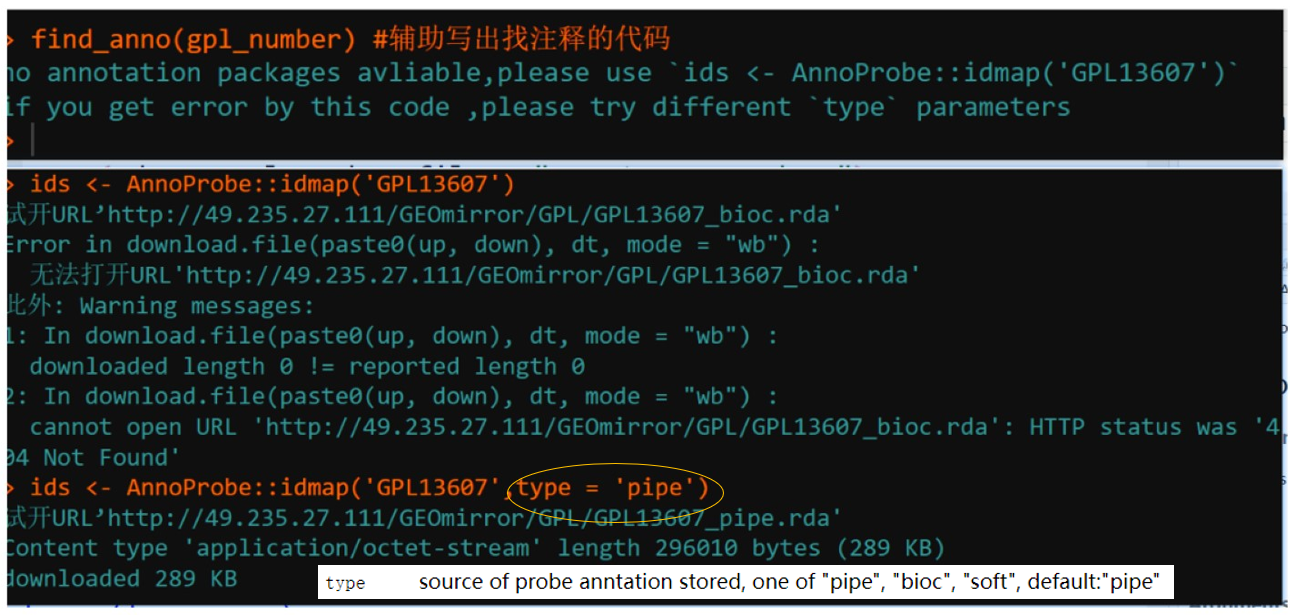
第三种情况显示no annotation avliable in Bioconductor and AnnoProbe则要去GEO网页上看GPL表格里找啦。
四种方法
- 方法1 BioconductorR包(最常用,已全部收入find_anno里面,不用看啦)
if(F){
gpl\_number #看看编号是多少
#http://www.bio-info-trainee.com/1399.html #在这里搜索,找到对应的R包
library(hgu133plus2.db)
ls("package:hgu133plus2.db") #列出R包里都有啥
ids <- toTable(hgu133plus2SYMBOL) #把R包里的注释表格变成数据框
}- 方法2 读取GPL网页的表格文件,按列取子集 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570




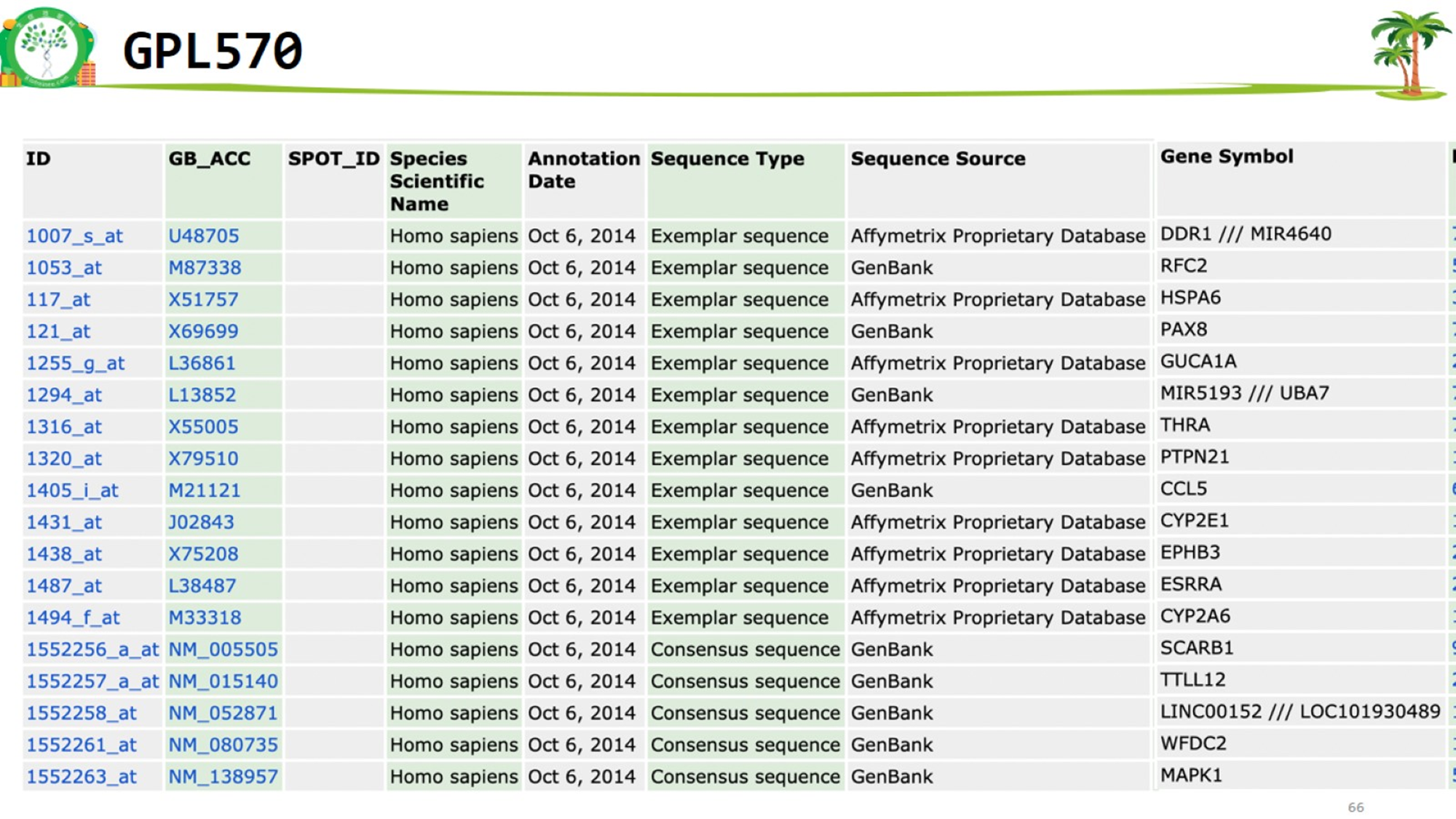
代码下载
#获取表格下载链接
get_gpl_txt(gpl_number)如何读取表格并提取子集,以GPL28098为例
#读取表格
a = data.table::fread("GPL28098.txt",data.table = F) # 提示丢了一行,所以换个读取函数
b = read.delim("GPL28098.txt",check.names = F,skip = 33) # 打开发现前33行是注释,跳过前33行
colnames(b)
ids = b[,c("ID" ,"SYMBOL")]
# 要改列名,后面的代码适应这两个列名
colnames(ids) = c("probe_id","symbol")问题:找不到symbol怎么办?
首先确认是不是基因表达芯片,可能是RNA芯片
然后看看别的列,基因名称可能包含在里面。比如GPL23126

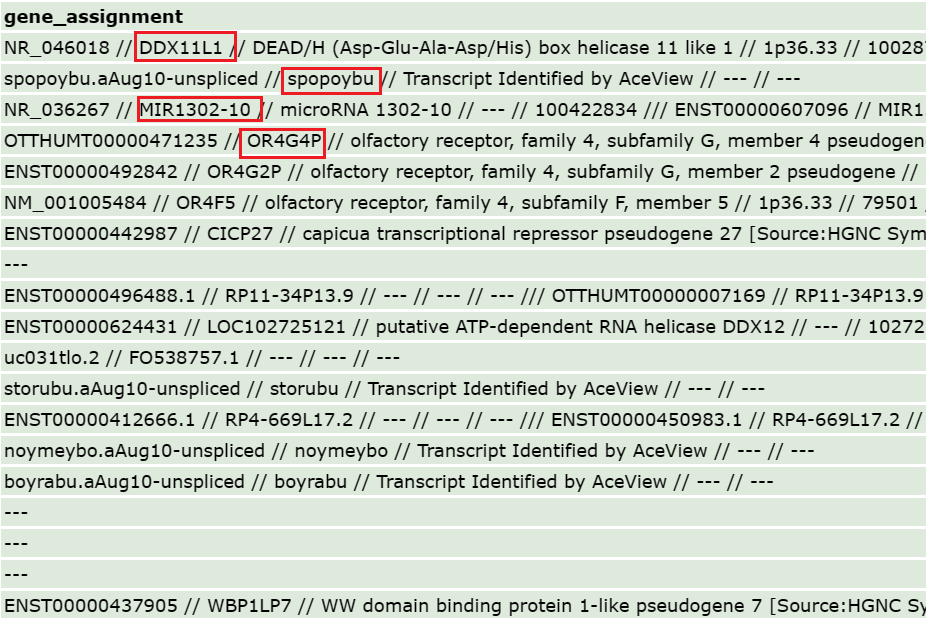
解决方法见小洁老师语雀 https://www.yuque.com/xiaojiewanglezenmofenshen/kzgwzl/sv262capcgg9o8s5?singleDoc# 《一个有点难的探针注释》
包含在ENTREZ_GENE_ID中
library(tinyarray)
find_anno("GPL30971")
get_gpl_txt("GPL30971") #网址复制到浏览器 下载到文件,放在工作目录下
f = data.table::fread("GPL30971.txt",data.table = F)
colnames(f)
ids = f[,c("ID","ENTREZ_GENE_ID")]
library(clusterProfiler)
library(org.Hs.eg.db)
e2s = bitr(ids$ENTREZ_GENE_ID,
fromType = "ENTREZID",
toType = "SYMBOL",
OrgDb = "org.Hs.eg.db")
ids = merge(ids,e2s,by.x = "ENTREZ_GENE_ID",by.y = "ENTREZID")
colnames(ids)
ids = ids[,-1]
ids = na.omit(ids)
colnames(ids) = c("probe_id","symbol")问题:网页里看symbol列是空的怎么办?
一般不影响,下载下来是有数据的
- 方法3 官网下载注释文件并读取
- 方法4 自主注释,了解一下
https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
不是所有芯片注释都能找到,不行就换个数据
保存运行结果
save(exp,Group,ids,file = "step2output.Rdata")画PCA图和热图
rm(list = ls())
load(file = "step2output.Rdata")
#输入数据:exp和Group
#Principal Component Analysis
#http://www.sthda.com/english/articles/31-principal-component-methods-in-r-practical-guide/112-pca-principal-component-analysis-essentials1.PCA 图------------------------------------------------------
dat=as.data.frame(t(exp))
library(FactoMineR)
library(factoextra)
dat.pca <- PCA(dat, graph = FALSE)
fviz_pca_ind(dat.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = Group, # color by groups
palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)2.top 1000 sd 热图----------------------------------------------------
g = names(tail(sort(apply(exp,1,sd)),1000)) #apply对每一行
n = exp[g,]
library(pheatmap)
annotation_col = data.frame(row.names = colnames(n),
Group = Group)
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col,
scale = "row", #按行标准化,只保留行内差别,不保留行间差别,会把数据范围缩放到大概-5~5之间
breaks = seq(-3,3,length.out = 100) #设置色带分布范围为-3~3之间,超出此范围的数字显示极限颜色
) 原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。