系统比较Seurat和scanpy版本之间、软件之间的分析差异
原创系统比较Seurat和scanpy版本之间、软件之间的分析差异
原创
作者,Evil Genius
标准的单细胞rna测序分析(scRNA-seq)工作流程软件括通过序列排列将原始读取数据转换为细胞基因计数矩阵,然后进行过滤、高变量基因选择、降维、聚类和差异表达分析等分析。Seurat和Scanpy是实现这种工作流的最广泛使用的软件,通常被认为是实现类似的单个步骤。
下面我们就需要比较一下软件之间、以及不同版本之间的数据分析差异。
单细胞rna测序(scRNA-seq)是一种强大的实验方法,为基因表达分析提供细胞分辨率。随着scRNA-seq技术的广泛应用,分析scRNA-seq数据的方法也越来越多。然而,尽管已经开发了大量的工具,但大多数scRNA-seq分析都是在两种分析平台之一进行的:Seurat或Scanpy。表面上,这些程序被认为实现了分析相同或非常相似的工作流程:scRNA-seq结果计算分析的第一步是将原始读取数据转换为细胞基因计数矩阵X,其中输入Xig是细胞i表达的基因g的RNA转录本的数量。通常,细胞和基因被过滤以去除质量差的细胞和最低表达的基因。然后,将数据归一化以控制无意义的可变性来源,如测序深度、技术噪声、库大小和批处理效果。然后从归一化数据中选择高度可变基因(hvg)来识别感兴趣的潜在基因并降低数据的维数。随后,基因表达值被缩放到跨细胞的平均值为0,方差为1**。这种缩放主要是为了能够应用主成分分析(PCA)来进一步降低维数,并提供有意义的嵌入来描述细胞之间的可变性来源。然后通过k近邻(KNN)算法传递细胞的PCA嵌入,以便根据细胞的基因表达描述细胞之间的关系。KNN图用于生成无向共享最近邻(SNN)图以供进一步分析,最近邻图被传递到聚类算法中,将相似的单元分组在一起。图(s)也用于进一步的非线性降维,使用t-SNE或UMAP在二维中图形化地描绘这些数据结构。最后,通过差异表达(DE)分析鉴定cluster特异性marker基因,其中每个基因的表达在每个cluster与所有其他cluster之间进行比较,并通过倍比变化和p值进行量化。
Seurat是2015年用R语言编写的,在生物信息学领域特别受欢迎;它是第一个全面的scRNA-seq分析平台之一。Scanpy是2017年继Seurat之后开发的一个基于python的工具,提供了一组类似的特性和功能。这两个工具都有广泛的运用。Seurat和Scanpy之间的选择通常归结为用户的编程偏好和他们的scRNA-seq数据分析项目的具体要求。
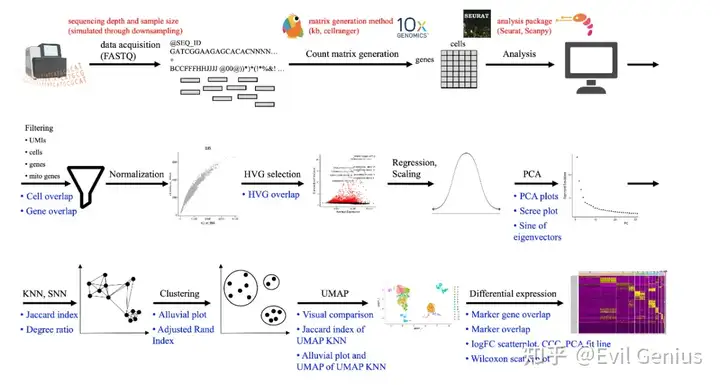
Seurat和Scanpy的输入由一个基因计数矩阵组成,通常是cellranger生成的矩阵。
一个“标准的”scRNA-seq实验需要花费数千美元,具体价格在很大程度上受数据大小的影响。虽然由于不同方法之间的差异,很难提供确切的成本,但据估计,一个典型的测序试剂盒的成本大约在数百到数千美元之间,测序成本每百万次读取5美元。对于高质量数据,每个cell的必要读取次数取决于实验的背景,但作为一个例子,cell Ranger通常建议其v3技术每个cell 20,000对reads,其v2技术每个cell 50,000对reads。样品制备也有很大的成本,通常需要宝贵的患者样本,或维持细胞或动物系数月至数年,为实验分析做准备。一个标准的10x Genomics scRNA-seq实验序列数千万到数十亿的reads,根据环境的不同,推荐的细胞计数范围为500-10k+。这些估计没有考虑额外的成本,包括人工、实验设置和后续分析。
生物信息学数据分析中一个典型的隐含假设是,软件和版本之间的选择应该对结果的解释几乎没有影响。然而,在软件或版本之间观察到相当大的可变性,即使在执行其他类似或看似相同的分析时也是如此。这里研究的目的是量化标准scRNA-seq pipeline在软件之间(即,Seurat与Scanpy)和同一软件的多个版本之间(即,Seurat v5与v4, Scanpy v1.9与v1.4, Cell Ranger v7与v6)的变异性。
Seurat和Scanpy在默认的scnaseq工作流中显示出相当大的差异
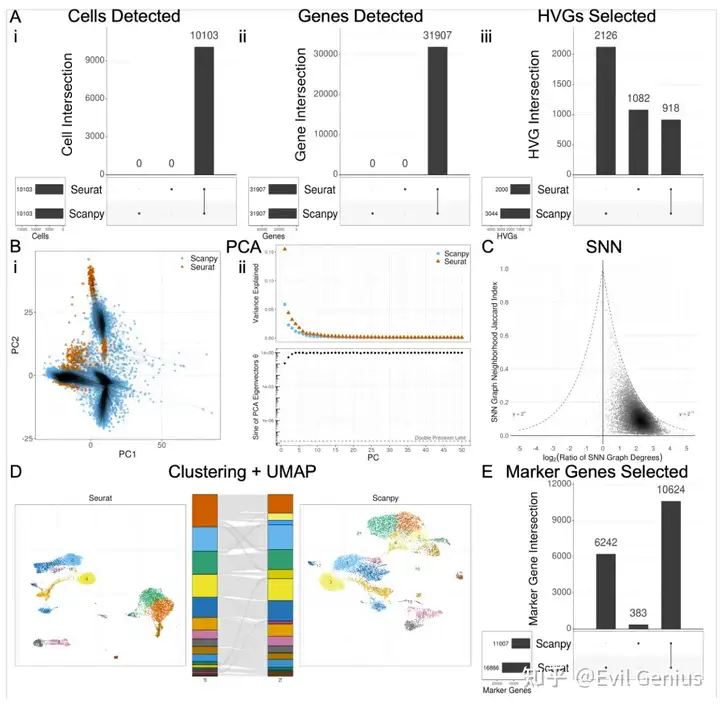
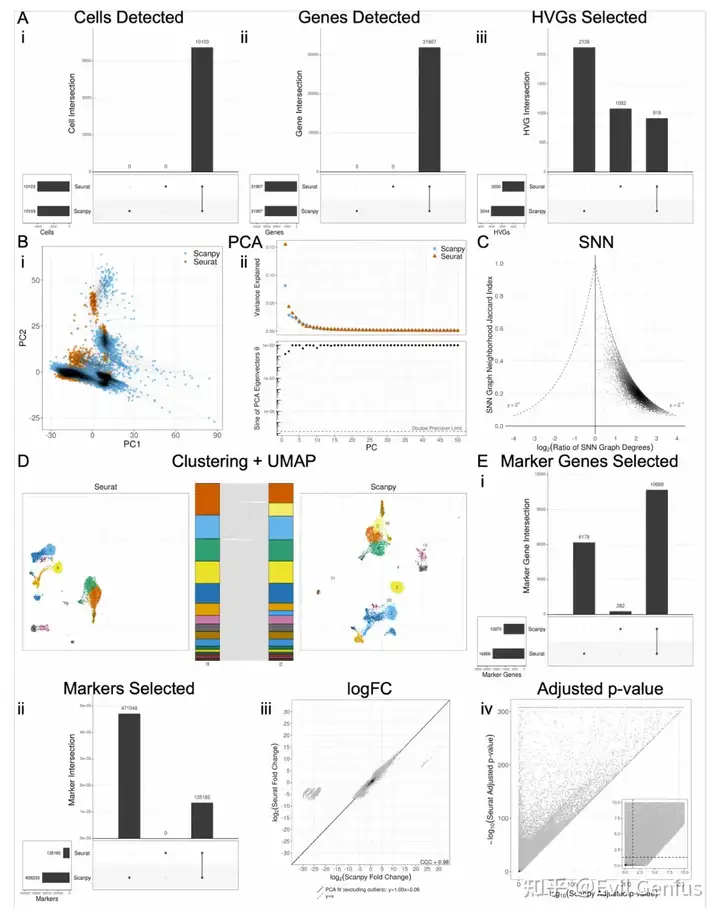
下图显示了使用PBMC 10k数据集与默认设置比较Seurat v5.0.2和Scanpy v1.9.5的结果,展示了“标准”单细胞RNA-seq工作流的两种实现之间的典型可变性。

在筛选UMIs、细胞最小基因数、基因最小细胞数和最大线粒体基因含量后,不同软件之间的细胞或基因过滤没有差异。此外,给定相同的矩阵作为输入,Seurat和Scanpy也以相同的方式处理日志规范化,产生等效的输出。然而,HVG选择的默认算法产生了差异,Jaccard index(两组之间差异基因的交集/并集)为0.22。This difference could be resolved either by selecting the “seurat v3” flavor for Scanpy or the “mean.var.plot” algorithm for Seurat。
PCA分析开始观察到更多的差异,使用默认参数运行时也会产生不同的结果。PCA图显示PC1-2空间中每个细胞的绘制位置存在明显差异,尽管图的大致形状保持不变。Scree图也显示出差异,最明显的是第一个PC解释的方差比例相差0.1。PCA的变化都可以通过HVG设置标准化来解决,并相应地调整PCA。
接下来,这些软件在SNN图的生成上有很大的不同。每个细胞的每个邻域的内容和大小都有很大差异。Seurat和Scanpy的每个细胞邻域间的Jaccard指数中位数为0.11,median degree ratio(Seurat/Scanpy)量级为2.05。每个函数的degree ratio几乎总是大于1,这表明Seurat在默认情况下比Scanpy给出更多的高连接SNN图。考虑到SNN图的点在所有degree ratio中相对均匀地分布在0和最大潜在Jaccard指数之间,似乎不是简单的度差驱动低平均Jaccard指数。在生成SNN图时对函数参数进行对齐,degree ratio没有质的提高,但中位Jaccard指数有轻微的提高。
使用默认设置的聚类也会导致输出的差异,即使在调整函数参数和输入SNN图时,Seurat和Scanpy也证明了Louvain聚类的差异,但在Leiden算法的实现中是相同的。
UMAP图在视觉上显示了局部和邻近cluster形状的一些差异,即使在控制全局移动或旋转的情况下。比较由这些UMAP数据构建的KNN图的邻域相似性,发现邻域重叠较差,随着函数参数和先前输入之间的相似性对齐,邻域重叠会适度改善。对这些由UMAP导出的KNN图进行Leiden聚类和随后的UMAP绘图,发现软件之间的UMAP绘图的一般特征保持不变,但仍然存在一些相当大的不可调和的差异。
在DE分析中,Seurat和Scanpy的显著性差异基因的Jaccard指数重合为0.62(即,所有聚类的基因总数调整后p值< 0.05),但Seurat的显著性marker基因比Scanpy多约50%。显著marker基因的差异是软件间默认设置的一些差异的结果。首先,每个软件分别实现Wilcoxon功能,Seurat需要tie校正,而Scanpy默认情况下省略tie校正。此外,每个软件在默认情况下调整p值不同- Seurat与Bonferroni multiple testing correction,而Scanpy与Benjamini-Hochberg多重测试校正。最后,Seurat在默认情况下,在执行Wilcoxon秩和检验之前,通过p值、每组拥有该基因的细胞百分比和对数倍变化(logFC)过滤marker;Scanpy在不调用其他函数的情况下不会执行这种类型的过滤。将Scanpy的过滤参数和聚类设置为与Seurat相同(filter:tie-correction, Bonferroni correction)进行DE分析,将marker基因重叠的Jaccard指数提高到0.73,提供相同的聚类分配进一步将Jaccard指数提高到0.99。其余1%的基因由于logFC计算的差异而存在差异。将方法设置为类似于Scanpy(没有过滤,Benjamini-Hochberg),使Jaccard指数恶化到0.38,因为无法去掉Seurat中的tie校正。
当对齐cluster信息时,可以执行进一步的DE分析,比较每个cluster中每个基因的表达水平差异。除了比较所有聚类中显著marker基因的外,还可以比较marker之间的相似性(即DE分析后每个聚类的基因)。Scanpy缺乏过滤意味着Scanpy包括所有cluster中的所有基因,即使该基因在该cluster中表达最少或无差异;而Seurat,默认情况下,根据logFC, p值和reference中表达基因的细胞数量,每个cluster只包含很小比例的基因。对Scanpy应用类似的阈值处理大大减少了这个问题,由于Scanpy缺少过滤,它将Jaccard指数从0.22提高到0.92。然而,这仍然不能完全调整差异。
Seurat和Scanpy计算logFC的方式也不同。比较各组间相似基因的一致性相关系数(CCC)为0.98,PCA拟合线斜率为1,表明各组间具有较强的相关性。简而言之,CCC衡量两个变量在相关性和方差方面的一致性。然而,通过观察logFC值的散点图,可以发现大量值之间存在显著差异。具体来说,在少数情况下(135185个marker中的4109个),Scanpy预测cluster中某个基因的logFC接近±30,而Seurat预测的logFC接近0。
在调整后的p值方面,Seurat和Scanpy之间也存在差异。对于默认的函数参数,Seurat预测的p值要么小于或类似于Scanpy,但不会大得多。大多数p值接近最大值1,但存在很大程度的变异性。相当多的p值远离y=x线,包括Seurat低于1e-50但Scanpy接近1的差异基因。20%的差异基因在软件之间的p值在p=0.05阈值上翻转,并且在两个方向上翻转相当均匀(即仅在Seurat中显著,或仅在Scanpy中显著)。当函数参数像Seurat一样对齐时,几乎所有调整后p值的差异都消失了。然而,由于在Seurat /presto的Wilcoxon秩和计算中缺乏切换校正的能力,这些差异无法与类似scanpy的函数参数相协调。
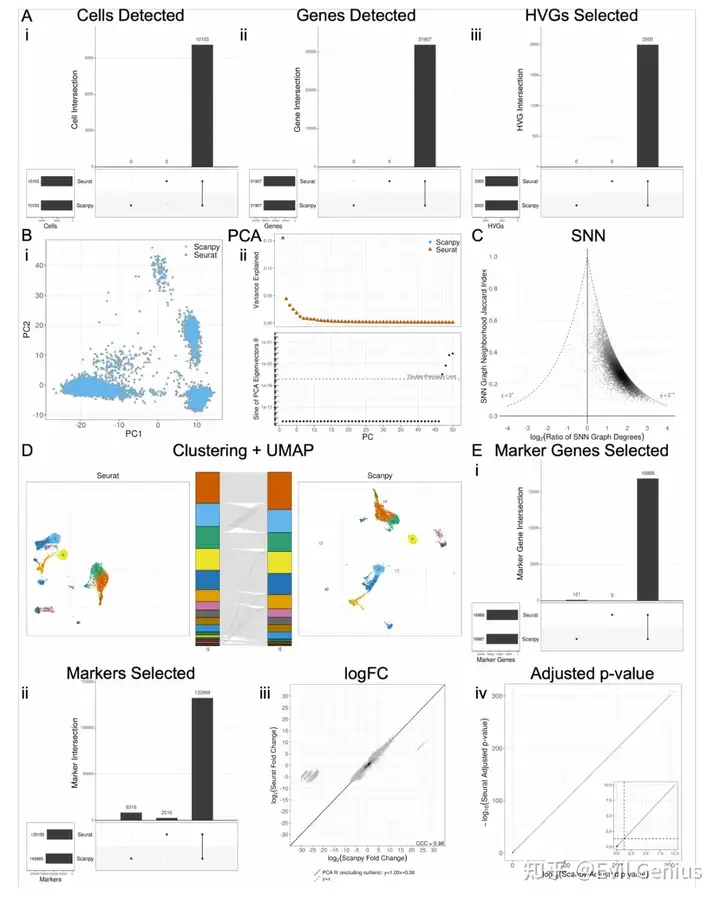
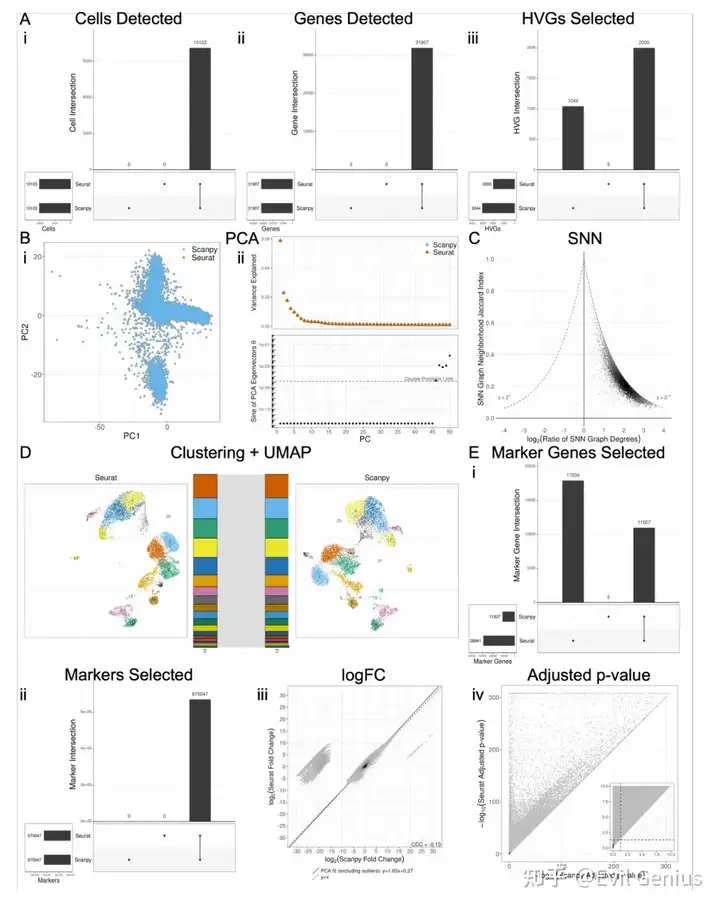
Seurat和Scanpy的比较表明,在某些情况下,程序结果是可以调和的,但并非总是如此。函数之间有三种可能的对齐方式:默认对齐、匹配函数参数时对齐、不兼容对齐。下表显示了每个函数在这些类中的分类。表1 (Seurat)和2 (Scanpy)详细介绍了分析函数名的每个步骤、默认参数、尽可能与其他软件匹配所需的参数以及该软件的唯一参数。

下采样比较
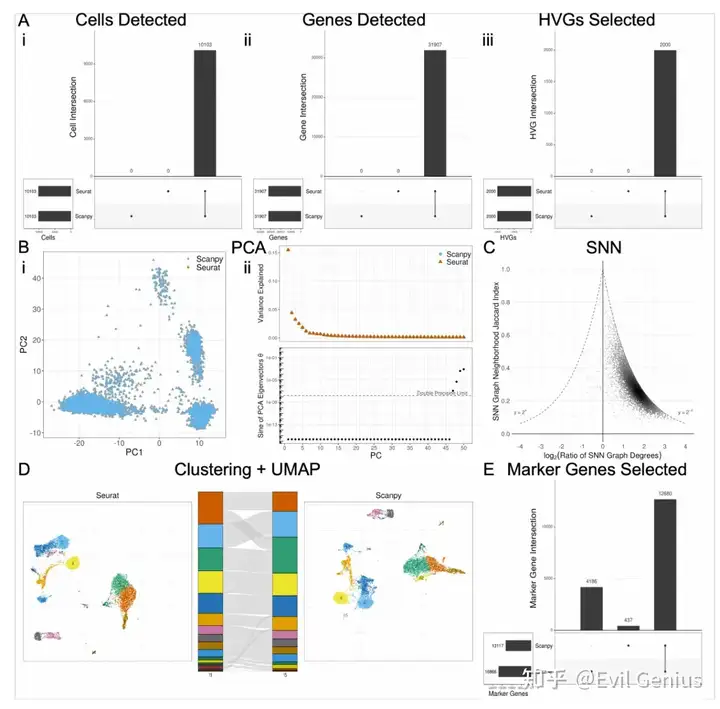
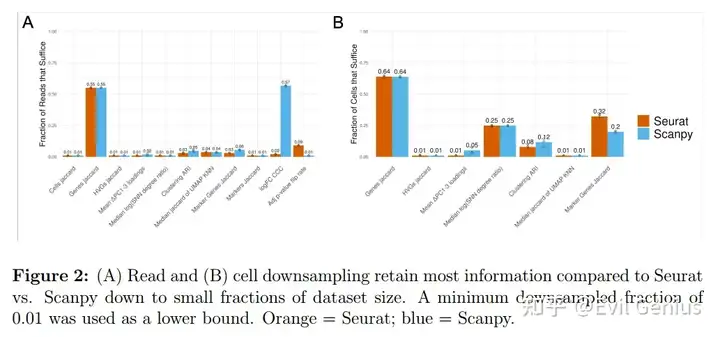
考虑到软件之间引入的可变性,一个自然的问题是如何对这些差异的大小进行基准测试。为此,在生成过滤UMI矩阵之前,模拟reads和细胞的下采样,并比较了沿下采样分数梯度引入的差异与全尺寸数据。我们使用各自软件的默认函数参数执行分析的每个步骤,并且在每个步骤之前不对齐输入数据,除了DE分析的那些步骤,这些步骤需要在每个步骤之前对齐输入,以便比较相同cluster中的差异基因统计(差异基因选择,logFC和调整的p值)。对于分析的每一步,除了生成如图1所示的所有图外,我们还选择了一个单一的数字指标,该指标将捕获组间差异的程度,如下所示:
? Cell filtering: Jaccard index of cell sets ? Gene filtering: Jaccard index of gene sets ? HVG selection: Jaccard index of HVG sets ? PCA: Mean of difference in corresponding PC loadings PCs 1-3 ? KNN/SNN: Median magnitude of log of SNN degree ratio ? Clustering: ARI ? UMAP: Median Jaccard index of UMAP-derived KNN neighborhoods across all cells ? Marker gene selection: Jaccard index of significant marker gene sets ? Marker selection: Jaccard index of marker sets ? logFC: CCC ? Adjusted p-value: Fraction of adjusted p-values that flipped across the p=0.05 threshold between conditions
对reads和细胞进行降采样的最不稳健的步骤是基因选择;然而,考虑到hvg和差异基因与下采样部分的全尺寸数据集的相似性,似乎基因集的差异主要在于不太重要的基因。
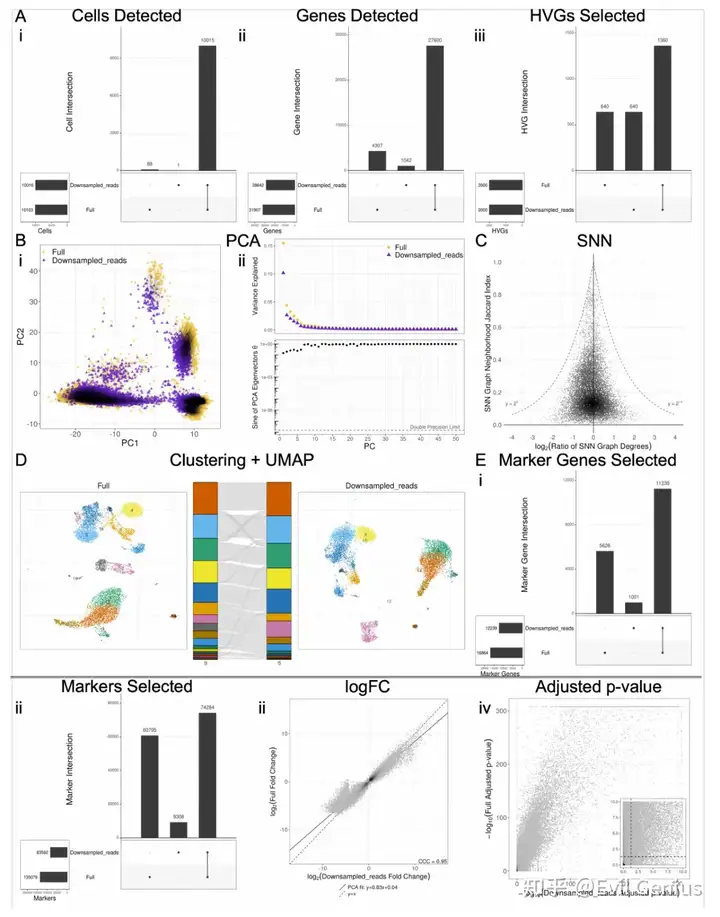
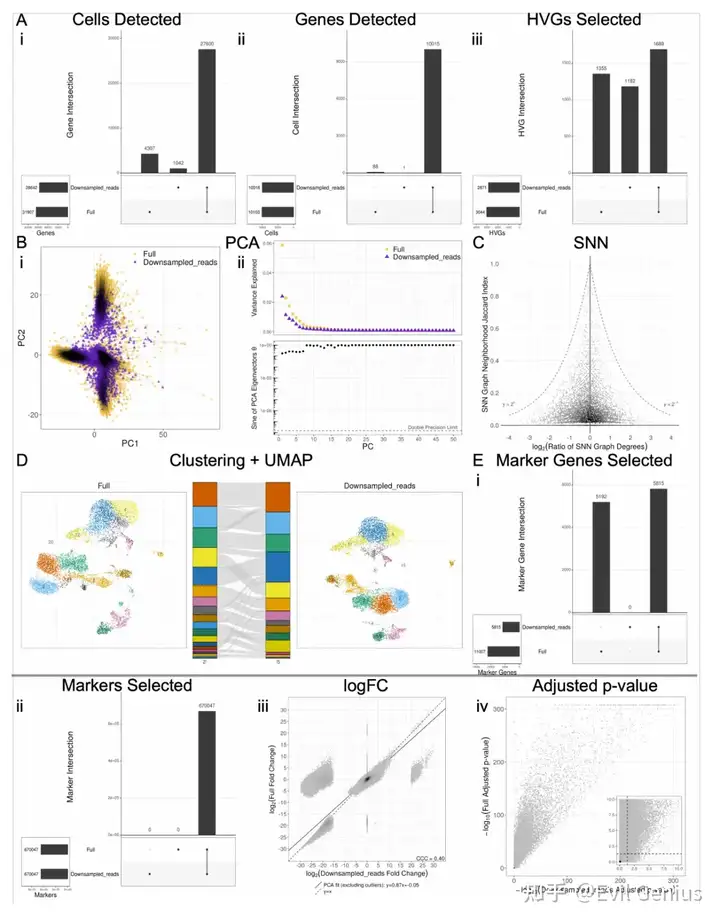
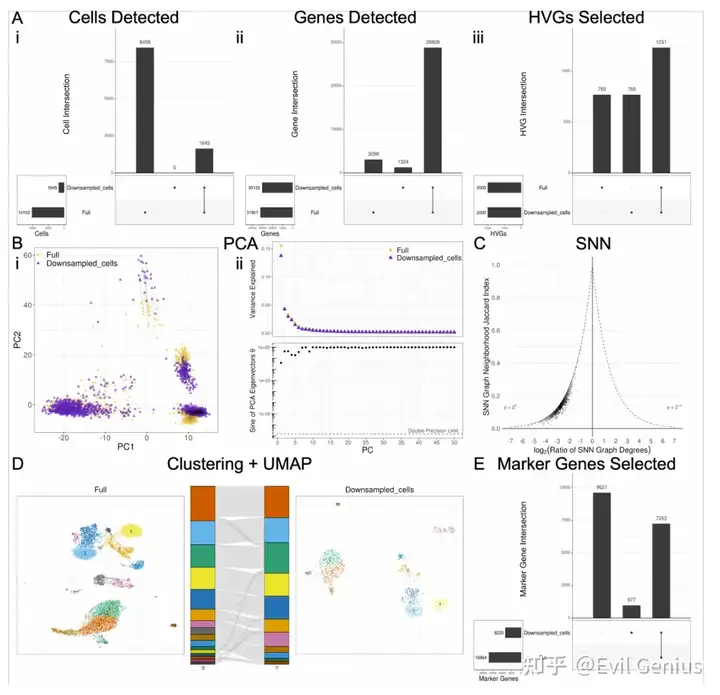
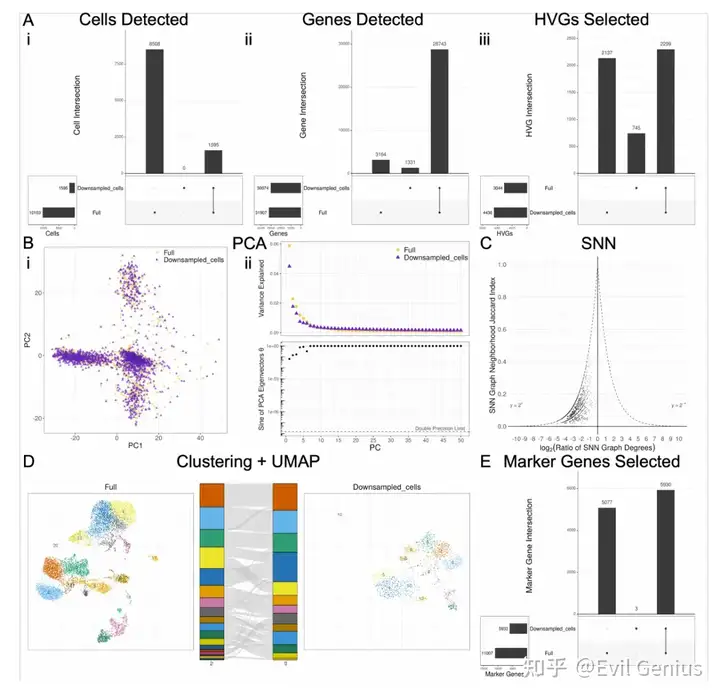
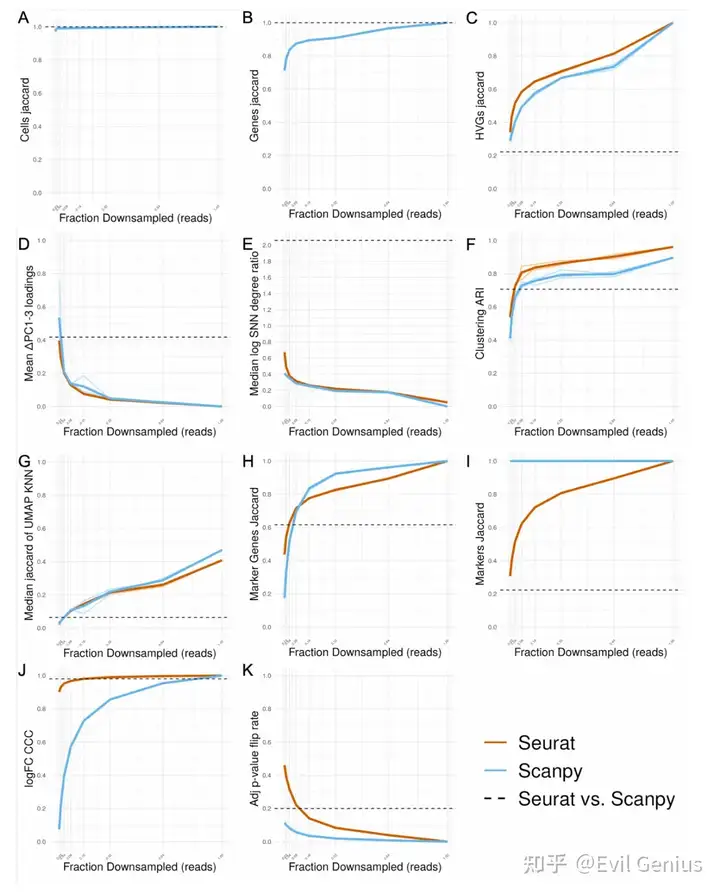
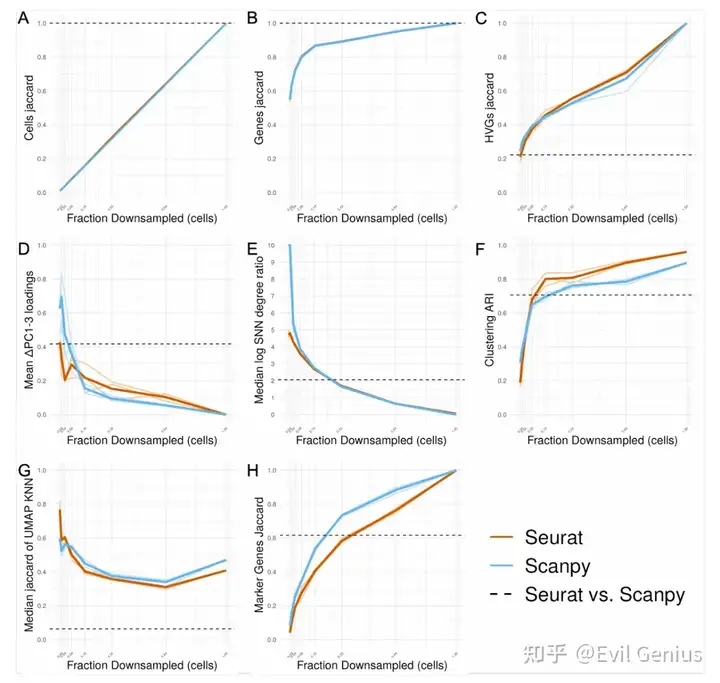
版本控制对DE分析具有重要意义
除了软件选择(例如,Seurat vs. Scanpy)之外,软件版本也可以在结果的解释中发挥作用。将Seurat v5与v4进行比较,在重要差异基因、marker和logFC估计值集方面存在相当大的差异。logFC计算的差异源于不同版本间伪计数应用程序的变化。Marker选择的差异完全来自于logFC计算和过滤参数的差异。将Scanpy v1.9与较早的v1.4进行比较还揭示了重要marker基因和marker list的巨大差异,这是由于删除了不同版本之间的marker过滤。这些版本之间的logFC计算和调整后的p值没有差异。比较使用默认设置的Cell Ranger软件v7和Cell Ranger v6生成的计数矩阵也揭示了所有DE指标之间的差异。跨Cell Ranger版本的分析显示,pipeline的所有步骤都存在相当大的差异。
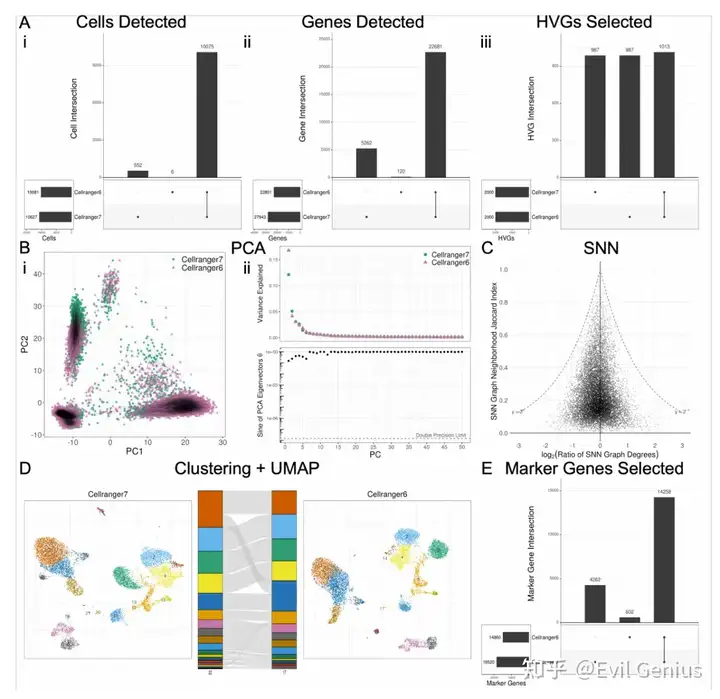
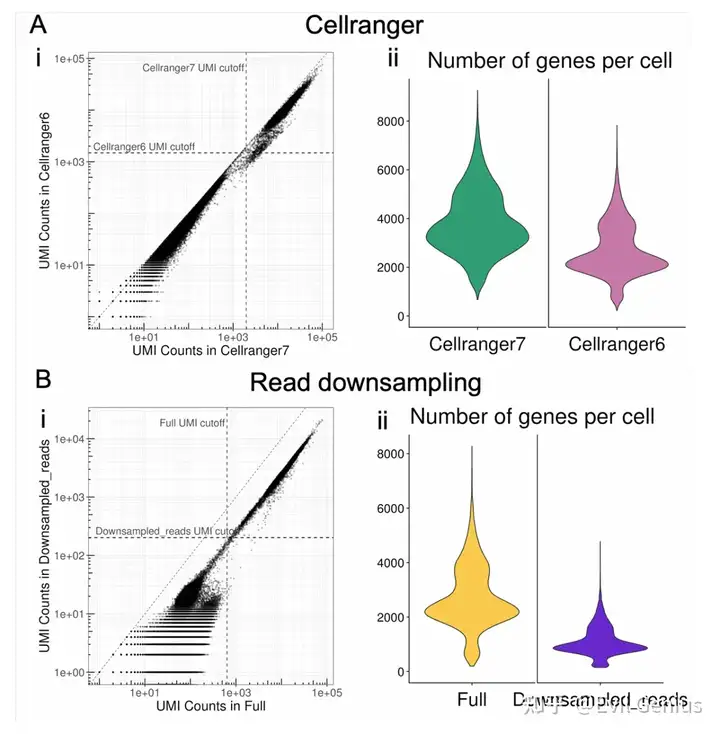
这些命令之间的主要区别在于v7中默认包含基因计数矩阵中的内含子计数,而v6中默认排除内含子计数。这种区别对UMI过滤和每个细胞的基因小提琴图有影响,与cell Ranger v6有更多的限制。

Random seeds
涉及随机化影响的工作流步骤有近似knn搜索、Louvain/Leiden图形聚类和UMAP。为了对软件或数据大小之间的差异程度进行基准测试,我们使用相同的输入数据和软件选择运行这些步骤,只改变应用的随机种子。在相同的PCA输入条件下,相同算法间SNN邻域的Jaccard指数中位数和对数度比的变化(Annoy为0.85和0.05,umap-learn/PyNNDescent为1和0)远低于Seurat和Scanpy的0.27和1.61对数度比,表明软件之间的差异不能仅仅用随机性来解释。随机种子间Louvain聚类后的ARI为0.96,显著高于Seurat和Scanpy中Louvain实现间的ARI(0.85)。UMAP衍生的随机UMAP种子间KNN的Jaccard指数中值,Seurat为0.41,Scanpy为0.47,显著高于相同输入为0.21的Seurat和Scanpy之间的UMAP图。然而,对于Seurat和Scanpy,在随机UMAP种子中对相同数据进行Leiden聚类后的ARI为0.64,与Seurat和Scanpy计算的观察到的ARI相似,给定相同的PCA和SNN输入,UMAP为0.69。这表明,尽管在Seurat或Scanpy中随机种子之间生成的UMAP图与软件之间生成的UMAP图具有更高的相似性,但Leiden算法不能完全捕获这种相似性。
总结
Seurat和Scanpy在使用默认设置执行分析的方式上存在相当大的差异,这些差异只能通过调整函数参数来部分调和。这些差异相当于当降采样读数小于5%或降采样细胞小于20%时引入的可变性。计数矩阵生成和分析中涉及的软件的版本控制也会对下游分析产生影响,特别是在没有仔细考虑跨版本行为变化的情况下。为了在scRNA-seq分析中实现准确性和可重复性,必须进行一致的封装选择、深思熟虑的参数选择和有意的版本控制。
参考文章在The impact of package selection and versioning on single-cell RNA-seq analysis。
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。