Salmon — 兼具高效、精准及偏差感知的RNA-seq定量工具
Salmon — 兼具高效、精准及偏差感知的RNA-seq定量工具

工欲善其事必先利其器
前面介绍了alevin 的单细胞定量功能,见 Alevin — 更快的单细胞定量,其也只是Salmon软件的一个组成部分,今天我们就再来学习一下 Salmon 其最初的功能 —— 转录组定量
1Salmon
Salmon 是由Rob Patro领导的团队开发的一款高效的生物信息学工具,主要用于定量转录组数据中的转录本表达量。其特性如下:
- 速度与效率:Salmon 使用了一种被称为“准映射”(quasi-mapping)的技术,该技术可以快速准确地将读段(reads)直接映射到转录本上,大大加快了处理速度并降低了内存需求。
- 无偏估计:软件在估计转录本丰度时,可以有效地避免系统偏误,使用复杂的统计模型来校正样本制备和测序过程中的偏差。
- 灵活的输入格式支持:Salmon 支持多种类型的输入数据,包括但不限于FASTA和FASTQ格式,能够处理单端和配对端数据。
官网:https://combine-lab.github.io/salmon/
GitHub:https://github.com/COMBINE-lab/salmon?tab=readme-ov-file
编程语言:C++

Salmon
2发表文章
文章信息:Salmon provides fast and bias-aware quantification of transcript expression 期刊:Nature Methods 日期:2017年3月 作者 & 单位:Rob Patro & Stony Brook University DOI:https://doi.org/10.1038/nmeth.4197

3如何安装
Salmon 提供了多种安装方式。包括但不限于 二进制版本、源码构建、Docker镜像、Conda安装等。由于源码构建所需依赖过多,所以推荐使用二进制版本、Conda、Docker镜像安装
Conda安装(推荐)
由于Github 没有提供最新版的salmon的二进制版本,所以推荐使用Conda安装
conda create -n salmon
conda activate salmon
conda install salmon -y
4定量基本流程
- 索引构建:首先,需要为参考转录组构建索引。这个过程包括对所有转录本的序列进行预处理,从而为快速准映射(quasi-mapping)做好准备。
- 准映射:在索引构建完成后,Salmon 使用准映射技术将读段(reads)直接映射到转录本上。这种方法不需要完全的比对,而是快速地估计读段与转录本的相似度,显著提升了处理速度。
- 表达量估计:映射完成后,Salmon 会根据映射的读段计算每个转录本的表达量。这一步考虑了读段覆盖度、转录本长度和测序深度等因素。
- 偏差校正:在定量的过程中,Salmon 还会进行偏差校正,尝试消除由测序和样本制备中引入的技术偏差影响。
- 输出结果:最后,Salmon 输出转录本的表达量估计,通常是以TPM(每百万转录本碱基数)单位表示。
5最小化使用
salmon有两种定量模式
基于映射模式的定量
准备文件
GENCODE:https://www.gencodegenes.org/
##下载对应物种的转录本fa文件
wget -c https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/gencode.v45.transcripts.fa.gz
## 创建salmon索引
salmon index -i homo_gencode_transcript_index -k 31 --gencode -p 8 -t gencode.v45.transcripts.fa.gz
-i #指定索引的输出位置和名称
-k #设置用于构建准索引的k-mer的长度,默认为31
-p #设置调用线程,默认为2
-t #输入文件;转录本的fasta格式文件
--gencode #此选项表明输入的fasta文件是GENCODE格式的,会在第一个`|`字符处切分转录本名称
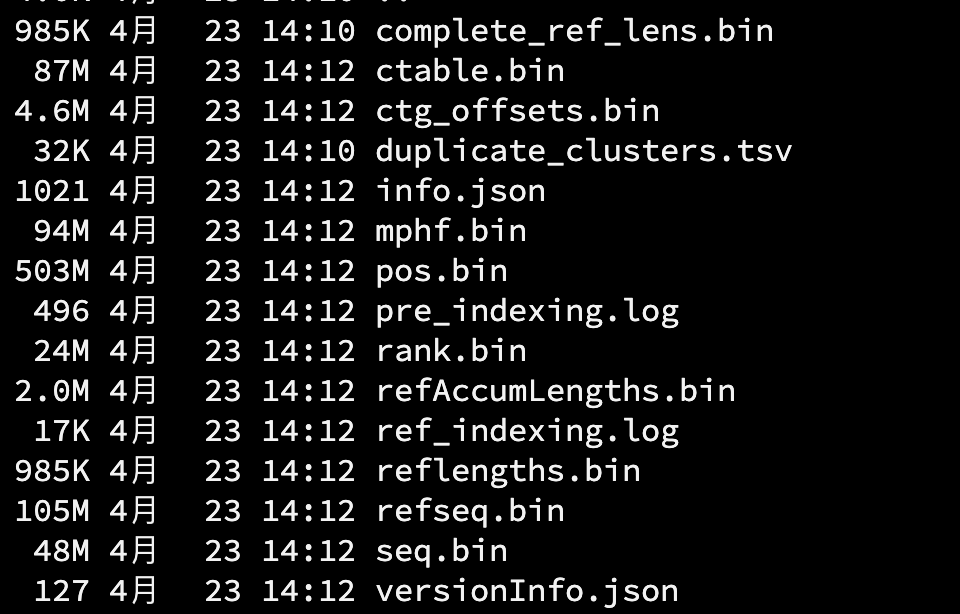
索引文件
当然,部分物种也可下载官方构建好的索引文件,见:http://refgenomes.databio.org/
定量
## 双端测序
## salmon quant -i transcripts_index -l <LIBTYPE> -1 reads1.fq -2 reads2.fq --validateMappings -o transcripts_quant
salmon quant -i ~/ref/homo/homo_gencode_transcript_index -l IU -p 8 -1 SRR8707539_1.fastq.gz -2 SRR8707539_2.fastq.gz -o ~/GSE128101/salmon_paired_out/
-i #指定索引文件位置
-l #指定库类型的格式字符串。这个参数告诉Salmon你的测序库是单端还是双端,以及测序的方向性 (注意参数顺序,-l 参数要放在reads文件之前),不清楚的话可以设为 “A”
-1 # reads 文件1
-2 # reads 文件2
-o #输出结果目录
-p #设置调用线程(默认256),建议8~12个线程
--validateMappings #早期版本的 Salmon 中,需要显式启用选择性对准来优化映射质量。在较新版本的Salmon中已被标记为弃用,并且没有效果。因为选择性对准(selective alignment)现在是Salmon的默认行为
## 单端测序
## salmon quant -i transcripts_index -l <LIBTYPE> -r reads.fq --validateMappings -o transcripts_quant
salmon quant -i ~/ref/mouse/mouse_gencode_transcript_index -l A -p 8 -r SRR8734841.fastq.gz -o ~/GSE128101/salmon_single_out/SRR8734841
-r # 指定单端样本
-l A #自动检测读段方向
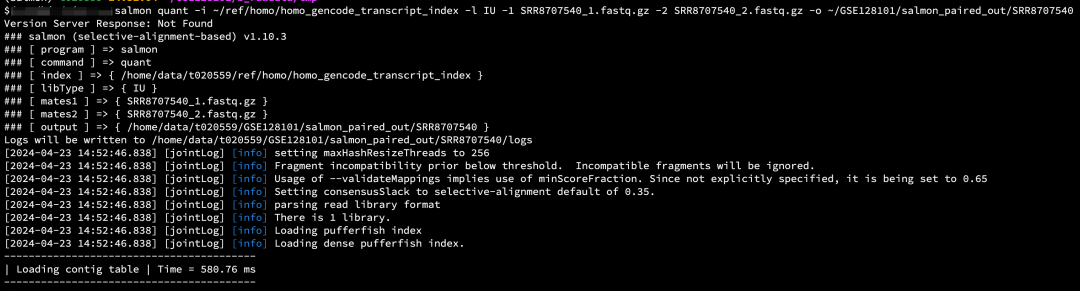
双端演示

单端演示
关于 -l 参数 库类型
对于单端测序
- A: 自动检测读段的方向
- U: 未指定方向性(未知或不相关)
对于双末端测序
- A:自动检测
- IU: 内向配对(inward),未指定方向性
- ISF: 内向配对,正向/正向
- ISR: 内向配对,反向/反向
- OSF: 外向配对,正向/正向
- OSR: 外向配对,反向/反向
- MSF: 匹配配对,正向/正向
- MSR: 匹配配对,反向/反向
I 代表内向(inward)配对,O 代表外向(outward)配对,M 代表匹配(matching)配对。S 代表正向(same),R 代表反向(reverse)

示意图
基于比对模式的定量
Salmon期望提供的对齐文件是针对与FASTA文件中给出的转录本进行的。这意味着读段应该直接对齐到转录本上,而不是整个基因组。这与一些其他工具(如Cufflinks)不同,后者是对整个基因组进行读段的对齐
## 比对
## hisat2 -p 4 -x ~/ref/homo/hisat2_trans_index/homo_gencode_v45 -1 SRR8707540_1_val_1.fq.gz -2 SRR8707540_2_val_2.fq.gz |samtools sort -@ 4 -o ~/GSE128101/hisat_trans_out/SRR8707540_sort.bam
## salmon基于比对模式的定量
## ./bin/salmon quant -t transcripts.fa -l <LIBTYPE> -a aln.bam -o salmon_quant
salmon quant -t ~/ref/homo/gencode.v45.transcripts.fa -l IU -p 8 -a ./SRR8707540_sort.bam -o ~/GSE128101/salmon_paired_out/SRR8707540_bam

基于比对模式
6结果文件
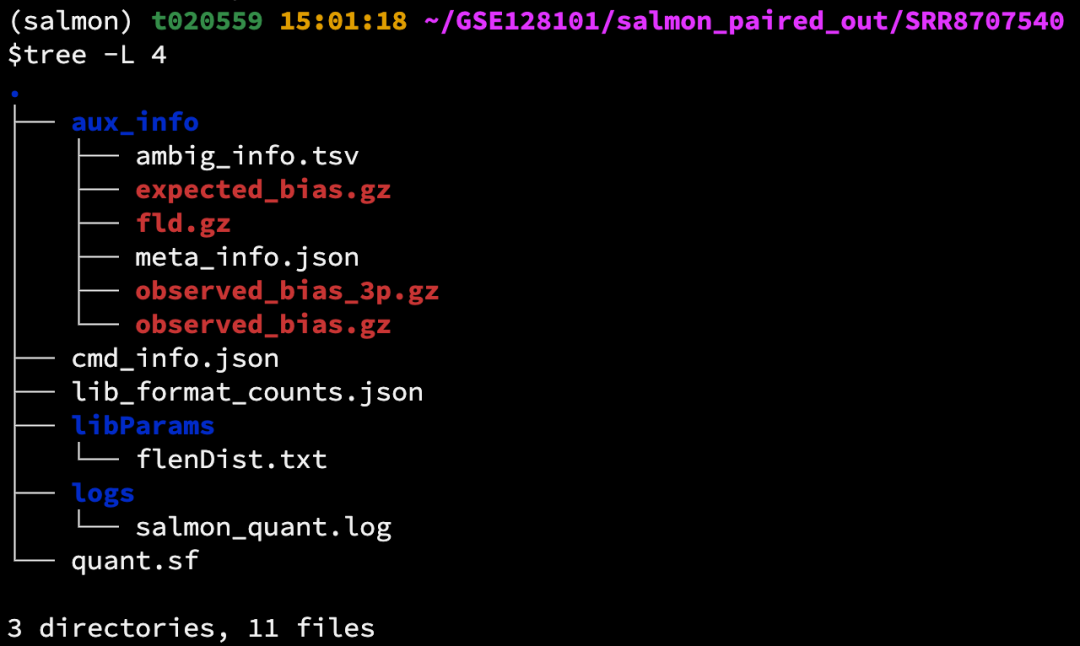
结果文件
主要结果
以制表符分割的定量文件 —— quant.sf
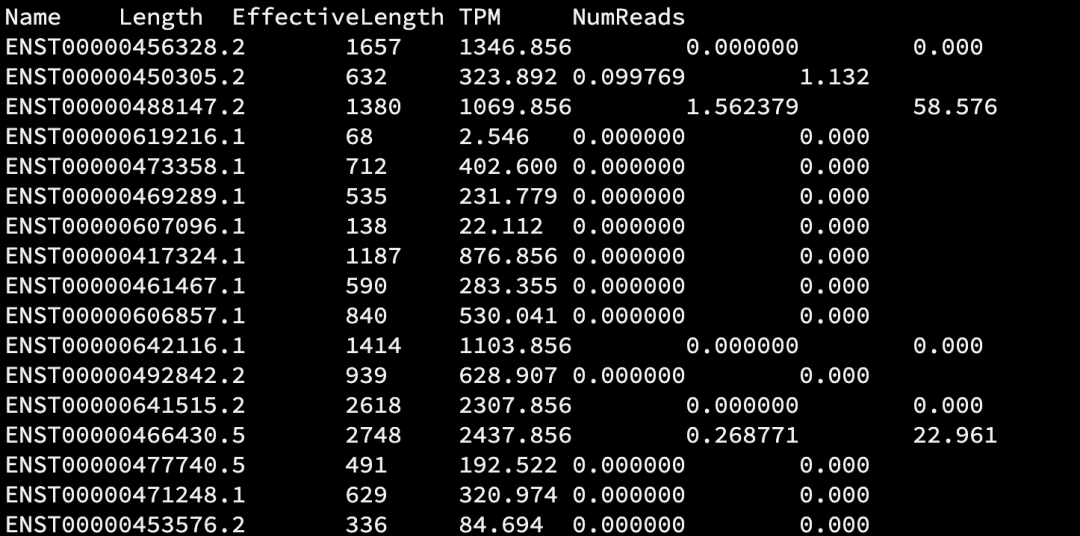
quant.sf
- Name(名称):输入转录本数据库(FASTA文件)中提供的目标转录本的名称
- Length(长度):目标转录本的长度,以核苷酸计算
- EffectiveLength(有效长度):这是计算得出的目标转录本的有效长度。它考虑了所有影响从该转录本采样片段的概率的因素,包括片段长度分布、序列特异性和GC片段偏差(如果这些因素被建模)
- TPM(每百万转录本数):Salmon估计的该转录本的相对丰度,单位为每百万转录本数(TPM)。TPM是推荐用于下游分析的相对丰度测量单位
- NumReads(读段数):Salmon估计的映射到每个被定量转录本的读段数量。这是一个“估计值”
更多参数信息详见:https://salmon.readthedocs.io/en/latest/salmon.html
参考:
- https://salmon.readthedocs.io/en/latest/salmon.html