单细胞分析|整合 scRNA-seq 和 scATAC-seq 数据
单细胞分析|整合 scRNA-seq 和 scATAC-seq 数据

引言
单细胞转录组学极大地提升了对细胞状态进行分类的能力,但要深入理解生物学现象,不能仅仅停留在对细胞群的简单列举上。随着新方法的不断涌现,用于测量细胞的不同状态,一个关键的挑战是如何将这些数据集整合起来,以便更全面地理解细胞的特性和功能。
举例来说,研究人员可能在同一生物体系中进行单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq)实验,并希望用同一套细胞类型标签来一致性地注释这两个数据集。这种分析尤其困难,因为scATAC-seq数据集的注释工作较为复杂,这不仅因为单细胞水平上收集的基因组数据较为稀疏,也因为scRNA-seq数据中缺少易于解释的基因标记。
在2019年由Stuart*, Butler*等人的研究中,引入了一种方法,用以整合来自同一生物体系的scRNA-seq和scATAC-seq数据集,并在本文[1]中展示了这些方法的应用。
分析要点
具体来说,展示了以下几项分析:
- 如何利用已注释的scRNA-seq数据集来标记scATAC-seq实验中的细胞
- 如何将scRNA-seq和scATAC-seq的细胞进行联合可视化(共同嵌入)
- 如何将scATAC-seq细胞映射到由scRNA-seq实验生成的UMAP空间中
本文大量使用了Signac软件包,这是一个新近开发的用于分析单细胞分辨率下收集的染色质数据集的工具,包括scATAC-seq数据。
采用了一个公开的约12,000个人类外周血单个核细胞(PBMC)的“多组学”数据集来演示这些方法,该数据集由10x Genomics公司提供。在这个数据集中,scRNA-seq和scATAC-seq的数据是在同一批细胞中同时获得的。为了本演示的目的,将这两个数据集视为来自两个独立的实验,并将它们进行了整合。由于这些数据原本是在相同细胞中测量的,因此为提供了一个基准,用以评估整合的准确性。在此强调,使用多组学数据集的目的是为了演示和评估,建议用户将这些方法应用于分别独立收集的scRNA-seq和scATAC-seq数据集。
加载数据并单独处理每种模式
外周血单个核细胞(PBMC)的多组学数据集可以通过10x Genomics公司获得。为了方便用户轻松地加载和探索数据,该数据集也被包含在的SeuratData包中。分别导入RNA和ATAC的数据,并假设这两组数据是分别在不同的实验中获得的。
library(SeuratData)
# install the dataset and load requirements
InstallData("pbmcMultiome")
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86)
library(ggplot2)
library(cowplot)
# load both modalities
pbmc.rna <- LoadData("pbmcMultiome", "pbmc.rna")
pbmc.atac <- LoadData("pbmcMultiome", "pbmc.atac")
pbmc.rna[["RNA"]] <- as(pbmc.rna[["RNA"]], Class = "Assay5")
# repeat QC steps performed in the WNN vignette
pbmc.rna <- subset(pbmc.rna, seurat_annotations != "filtered")
pbmc.atac <- subset(pbmc.atac, seurat_annotations != "filtered")
# Perform standard analysis of each modality independently RNA analysis
pbmc.rna <- NormalizeData(pbmc.rna)
pbmc.rna <- FindVariableFeatures(pbmc.rna)
pbmc.rna <- ScaleData(pbmc.rna)
pbmc.rna <- RunPCA(pbmc.rna)
pbmc.rna <- RunUMAP(pbmc.rna, dims = 1:30)
# ATAC analysis add gene annotation information
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevelsStyle(annotations) <- "UCSC"
genome(annotations) <- "hg38"
Annotation(pbmc.atac) <- annotations
# We exclude the first dimension as this is typically correlated with sequencing depth
pbmc.atac <- RunTFIDF(pbmc.atac)
pbmc.atac <- FindTopFeatures(pbmc.atac, min.cutoff = "q0")
pbmc.atac <- RunSVD(pbmc.atac)
pbmc.atac <- RunUMAP(pbmc.atac, reduction = "lsi", dims = 2:30, reduction.name = "umap.atac", reduction.key = "atacUMAP_")
现在绘制两种模式的结果。先前已根据转录组状态对细胞进行了注释。将预测 scATAC-seq 细胞的注释。
p1 <- DimPlot(pbmc.rna, group.by = "seurat_annotations", label = TRUE) + NoLegend() + ggtitle("RNA")
p2 <- DimPlot(pbmc.atac, group.by = "orig.ident", label = FALSE) + NoLegend() + ggtitle("ATAC")
p1 + p2

plot <- (p1 + p2) & xlab("UMAP 1") & ylab("UMAP 2") & theme(axis.title = element_text(size = 18))
ggsave(filename = "../output/images/atacseq_integration_vignette.jpg", height = 7, width = 12, plot = plot,
quality = 50)
识别 scRNA-seq 和 scATAC-seq 数据集之间的anchors
为了在单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq)实验之间找到相互关联的“锚点”,首先利用Signac软件包中的GeneActivity()函数,通过计算2kb启动子区域和基因体内的ATAC-seq测序计数,来估算每个基因的转录活性。
接下来,使用scATAC-seq数据得到的基因活性评分,与scRNA-seq中的基因表达量数据一起,作为典型相关性分析的输入。对scRNA-seq数据集中所有被鉴定为变异性高的基因进行这样的量化分析。
# quantify gene activity
gene.activities <- GeneActivity(pbmc.atac, features = VariableFeatures(pbmc.rna))
# add gene activities as a new assay
pbmc.atac[["ACTIVITY"]] <- CreateAssayObject(counts = gene.activities)
# normalize gene activities
DefaultAssay(pbmc.atac) <- "ACTIVITY"
pbmc.atac <- NormalizeData(pbmc.atac)
pbmc.atac <- ScaleData(pbmc.atac, features = rownames(pbmc.atac))
# Identify anchors
transfer.anchors <- FindTransferAnchors(reference = pbmc.rna, query = pbmc.atac, features = VariableFeatures(object = pbmc.rna),
reference.assay = "RNA", query.assay = "ACTIVITY", reduction = "cca")
通过标签转移注释 scATAC-seq 细胞
确定锚点之后,能够将单细胞RNA测序(scRNA-seq)数据集上的细胞类型注释应用到单细胞ATAC测序(scATAC-seq)的细胞上。这些注释保存在seurat_annotations字段里,并作为参考数据输入到refdata参数中。最终的输出结果会是一个矩阵,其中包含了每个ATAC-seq细胞的预测分类和置信度分数。
celltype.predictions <- TransferData(anchorset = transfer.anchors, refdata = pbmc.rna$seurat_annotations,
weight.reduction = pbmc.atac[["lsi"]], dims = 2:30)
pbmc.atac <- AddMetaData(pbmc.atac, metadata = celltype.predictions)
- 为什么会选用不同的(非默认设置的)参数值来进行降维和权重调整?
在执行FindTransferAnchors()函数时,通常会在单细胞RNA测序(scRNA-seq)数据集间进行数据转移时,将参考数据集的PCA(主成分分析)结构映射到查询数据集上。但是,在不同技术平台间进行数据转移时,发现典型相关分析(CCA)能更准确地捕捉到共享特征之间的相关性结构,因此在这里选择将降维方法设置为'cca'。
此外,在TransferData()函数中,默认情况下会利用相同的PCA映射结构来计算局部锚点邻域的权重,这些锚点对每个细胞的预测结果有影响。特别是在从scRNA-seq转移到scATAC-seq的过程中,会采用在ATAC-seq数据上通过计算LSI(潜语义索引)得到的低维空间来计算这些权重,因为这种方法能更好地反映ATAC-seq数据的内在结构特征。
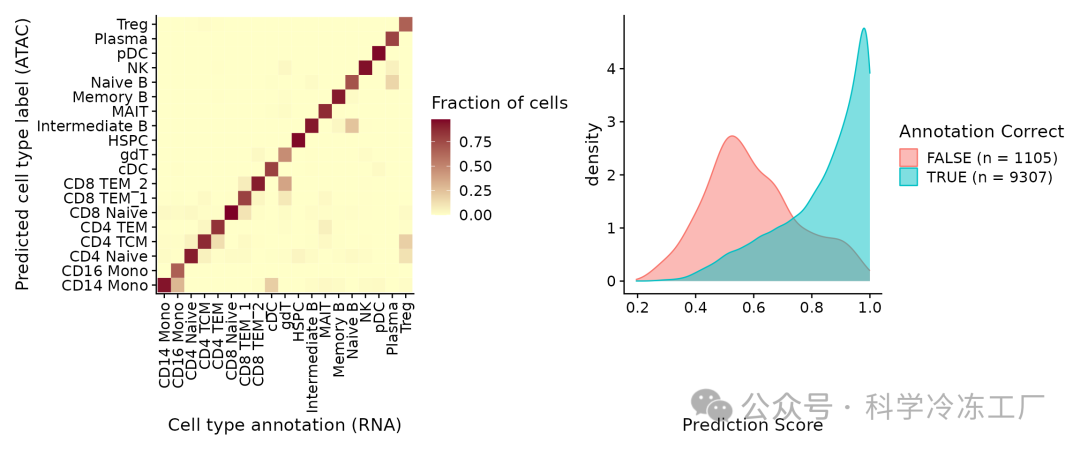
完成注释转移后,ATAC-seq的细胞获得了预测的细胞类型标签,这些标签源自scRNA-seq数据集,并存储在predicted.id字段中。鉴于这些细胞是通过多组学试剂盒进行测量的,还拥有一套真实的细胞类型标签,可以用来进行评估。可以观察到,预测得出的细胞类型标签与实际的标签极为相似。
pbmc.atac$annotation_correct <- pbmc.atac$predicted.id == pbmc.atac$seurat_annotations
p1 <- DimPlot(pbmc.atac, group.by = "predicted.id", label = TRUE) + NoLegend() + ggtitle("Predicted annotation")
p2 <- DimPlot(pbmc.atac, group.by = "seurat_annotations", label = TRUE) + NoLegend() + ggtitle("Ground-truth annotation")
p1 | p2

以本例而言,通过单细胞RNA测序(scRNA-seq)数据的整合,单细胞ATAC测序(scATAC-seq)的细胞类型注释正确预测的准确率可达约90%。另外,prediction.score.max字段用于衡量预测注释的不确定性。可以观察到,正确注释的细胞通常伴随着较高的预测分数(超过90%),而错误注释的细胞则通常伴随着显著较低的预测分数(低于50%)。此外,错误的分类往往涉及到非常相似的细胞类型,例如中间态B细胞与原始B细胞之间的区分。
predictions <- table(pbmc.atac$seurat_annotations, pbmc.atac$predicted.id)
predictions <- predictions/rowSums(predictions) # normalize for number of cells in each cell type
predictions <- as.data.frame(predictions)
p1 <- ggplot(predictions, aes(Var1, Var2, fill = Freq)) + geom_tile() + scale_fill_gradient(name = "Fraction of cells",
low = "#ffffc8", high = "#7d0025") + xlab("Cell type annotation (RNA)") + ylab("Predicted cell type label (ATAC)") +
theme_cowplot() + theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1))
correct <- length(which(pbmc.atac$seurat_annotations == pbmc.atac$predicted.id))
incorrect <- length(which(pbmc.atac$seurat_annotations != pbmc.atac$predicted.id))
data <- FetchData(pbmc.atac, vars = c("prediction.score.max", "annotation_correct"))
p2 <- ggplot(data, aes(prediction.score.max, fill = annotation_correct, colour = annotation_correct)) +
geom_density(alpha = 0.5) + theme_cowplot() + scale_fill_discrete(name = "Annotation Correct",
labels = c(paste0("FALSE (n = ", incorrect, ")"), paste0("TRUE (n = ", correct, ")"))) + scale_color_discrete(name = "Annotation Correct",
labels = c(paste0("FALSE (n = ", incorrect, ")"), paste0("TRUE (n = ", correct, ")"))) + xlab("Prediction Score")
p1 + p2
共同嵌入 scRNA-seq 和 scATAC-seq 数据集
除了能够在不同技术平台间传递细胞标签,还能够在同一个图表中同时展示单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq)的细胞分布。需要指出的是,这一步骤主要用于图形展示,并不是必须执行的。
通常情况下,在进行scRNA-seq与scATAC-seq数据集之间的整合分析时,主要集中于上述的标签传递工作。在下文中展示了如何进行细胞数据的共同嵌入,并再次强调,这仅用于示范,特别是在本案例中,scRNA-seq和scATAC-seq的数据实际上是从同一批细胞中获得的。
为了实现共同嵌入,会首先基于之前计算得到的锚点,对scATAC-seq细胞进行RNA表达量的“估算”,然后将这些数据集进行合并。
# note that we restrict the imputation to variable genes from scRNA-seq, but could impute the
# full transcriptome if we wanted to
genes.use <- VariableFeatures(pbmc.rna)
refdata <- GetAssayData(pbmc.rna, assay = "RNA", slot = "data")[genes.use, ]
# refdata (input) contains a scRNA-seq expression matrix for the scRNA-seq cells. imputation
# (output) will contain an imputed scRNA-seq matrix for each of the ATAC cells
imputation <- TransferData(anchorset = transfer.anchors, refdata = refdata, weight.reduction = pbmc.atac[["lsi"]],
dims = 2:30)
pbmc.atac[["RNA"]] <- imputation
coembed <- merge(x = pbmc.rna, y = pbmc.atac)
# Finally, we run PCA and UMAP on this combined object, to visualize the co-embedding of both
# datasets
coembed <- ScaleData(coembed, features = genes.use, do.scale = FALSE)
coembed <- RunPCA(coembed, features = genes.use, verbose = FALSE)
coembed <- RunUMAP(coembed, dims = 1:30)
DimPlot(coembed, group.by = c("orig.ident", "seurat_annotations"))

Reference
[1] Source: https://satijalab.org/seurat/articles/seurat5_atacseq_integration_vignette