多样本空间转录组学联合解卷积分析与跨样本转录相似性分析
原创多样本空间转录组学联合解卷积分析与跨样本转录相似性分析
原创
追风少年i
发布于 2024-05-06 14:28:03
发布于 2024-05-06 14:28:03
作者,Evil Genius
关于空间转录组的分析是呈现一个螺旋上升的过程。
在文章Cell | 空间转录组数据分析的潜力中提到空间转录组学方法产生三种不同但相互关联的数据类型:(1)图像数据,(2)表达数据,(3)空间方向和位置。当然这还是指的空间单样本,多样本的分析自然也会产生很多复杂的问题。
关于空间多样本的整合分析之前分享过,文章在空转第三节课多样本整合的补充
单细胞 & 空间整合去批次方法比较
单细胞 & 空间整合去批次方法比较(2)
空转第三节课多样本整合的补充2(python版本)
其中空间整合也涉及到上面三个组学数据的内容。其中细胞进化成组织区域并相互作用的过程取决于与周围环境的相互作用,因此ST技术自然保存的空间信息为增强对疾病进展和组织发育的理解提供了充足的条件。
目前限于空间的精度,单细胞空间联合分析的软件包括
CARD,BayesTME,STdeconvolve,Cell2location,DestVI,RCTD,EnDecon,SPOTlight,and UniCell
空间多样本分析需要解决的生物学问题
- 多样品ST数据集的生成,使数据集成和统计建模能够更可靠地比较、验证和识别空间调节的基因表达模式(例如:多样本ST允许在不同条件下(例如,敲除与野生型)或实验环境下(例如,治疗应答与无应答)更全面地研究基因表达空间动态)。
- 样品间的比较分析提供了对基因表达的空间调控的见解,揭示了在单样品ST分析中可能被忽视的空间集群和协调的基因模块。
那么我们的目标就是:提供工具用于多样本ST数据集的综合分析,其中填补的空白包括
- 多样本ST数据分析的无参考数据集反卷积方法。
- 样本内和样本间的信息共享,构建跨样本网络图
- 跨ST样本的生物信号矫正。
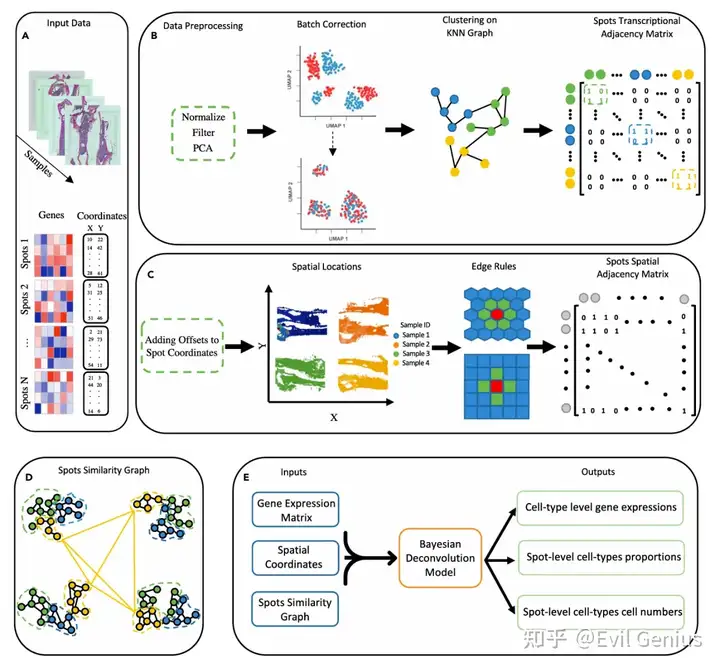
空间多样本分析框架
核心步骤如下
(1)构建基于表达的组织样本间批量效应校正后信息共享的点转录邻接矩阵; (2)构建点空间邻接矩阵,实现样本内物理相邻点之间的空间关联; (3)基于点转录矩阵和空间邻接矩阵构建点相似图; (4)基于贝叶斯层次模型的聚合点水平基因表达测量对来自不同细胞类型的信号的反卷积。
其中,Spot transcriptional adjacency matrix
前期处理类似单细胞的方法,归一化、特征选择和降维等,harmony去除批次并且计算转录相似性,聚类。
Spot spatial adjacency matrix
构造一个spot空间临近矩阵,即一个点的临近的6个点的转录信息。
多样本联合解卷积分析(Bayesian deconvolution model)
我们来看看具体的效果
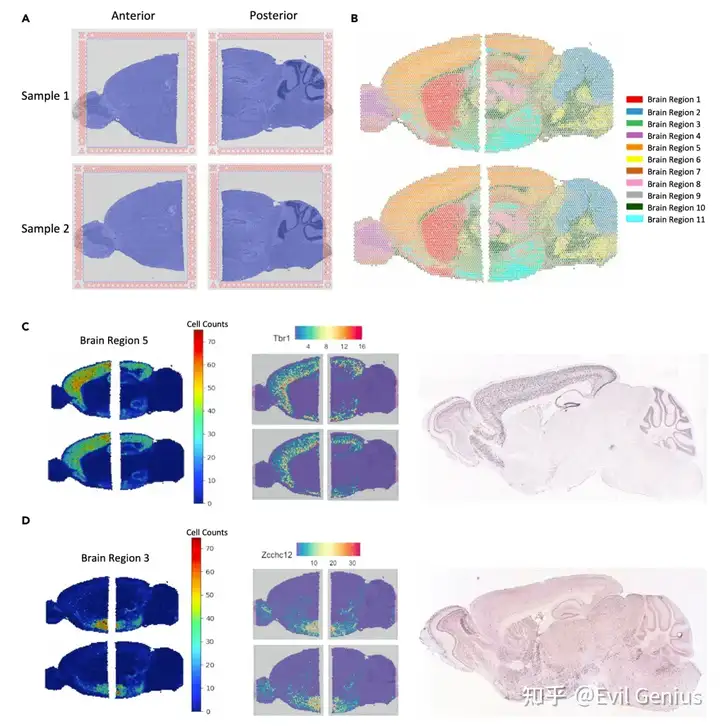
脑部联合分析
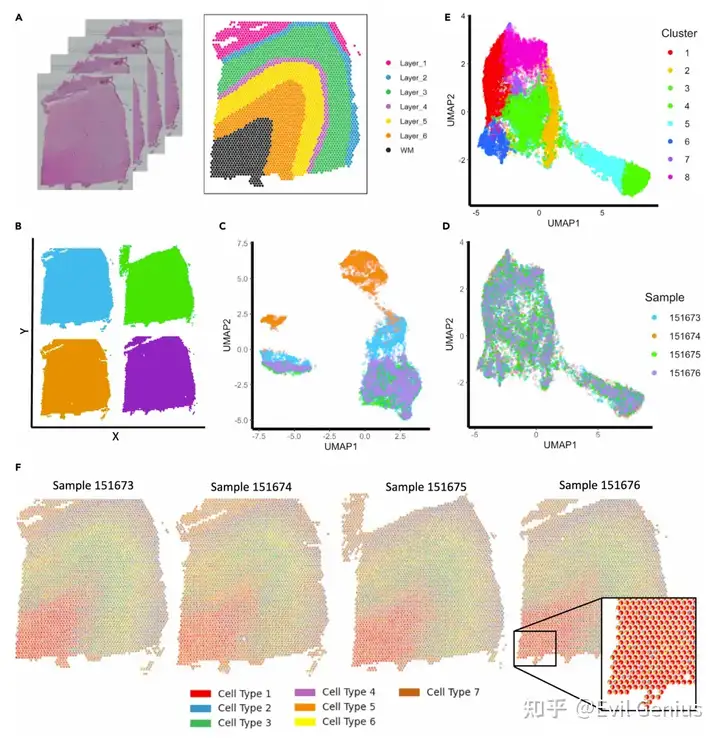
多样本联合解卷积分析
看一看示例代码
加载数据
suppressPackageStartupMessages({
library(ggplot2)
library(patchwork)
library(scater)
library(harmony)
library(BayesSpace)
})
set.seed(100)
matrix_dir="~/Documents/Spatial/mouse_brain/Sec1_Anterior/outs/"
sec1_anterior <- SpatialExperiment::read10xVisium(samples = matrix_dir,
type = "HDF5",
data = "filtered",load = T)
colData(sec1_anterior)$sample_id<-"Sec1_Anterior"
matrix_dir="~/Documents/Spatial/mouse_brain/Sec1_Posterior/outs/"
sec1_posterior <- SpatialExperiment::read10xVisium(samples = matrix_dir,
type = "HDF5",
data = "filtered",load = T)
colData(sec1_posterior)$sample_id<-"Sec1_Posterior"
matrix_dir="~/Documents/Spatial/mouse_brain/Sec2_Anterior/outs/"
sec2_anterior <- SpatialExperiment::read10xVisium(samples = matrix_dir,
type = "HDF5",
data = "filtered",load = T)
colData(sec2_anterior)$sample_id<-"Sec2_Anterior"
matrix_dir="~/Documents/Spatial/mouse_brain/Sec2_Posterior/outs/"
sec2_posterior <- SpatialExperiment::read10xVisium(samples = matrix_dir,
type = "HDF5",
data = "filtered",load = T)
colData(sec2_posterior)$sample_id<-"Sec2_Posterior"
rowData(sec1_anterior)$is.HVG = NULL
rowData(sec1_posterior)$is.HVG = NULL
rowData(sec2_anterior)$is.HVG = NULL
rowData(sec2_posterior)$is.HVG = NULL
for(i in 1:nrow(colData(sec1_anterior))){
colData(sec1_anterior)@rownames[i]<-paste0("Sec1_Ant_",colData(sec1_anterior)@rownames[i])
}
for(i in 1:nrow(colData(sec1_posterior))){
colData(sec1_posterior)@rownames[i]<-paste0("Sec1_Post_",colData(sec1_posterior)@rownames[i])
}
for(i in 1:nrow(colData(sec2_anterior))){
colData(sec2_anterior)@rownames[i]<-paste0("Sec2_Ant_",colData(sec2_anterior)@rownames[i])
}
for(i in 1:nrow(colData(sec2_posterior))){
colData(sec2_posterior)@rownames[i]<-paste0("Sec2_Post_",colData(sec2_posterior)@rownames[i])
}
#Combine into 1 SCE and preprocess
sce.combined = cbind(sec2_anterior, sec1_anterior, sec2_posterior, sec1_posterior, deparse.level = 1)
sce.combined = spatialPreprocess(sce.combined, n.PCs = 50, n.HVGs=2000,assay.type="logcounts") #lognormalize, PCA
sce.combined = runUMAP(sce.combined, dimred = "PCA")
colnames(reducedDim(sce.combined, "UMAP")) = c("UMAP1", "UMAP2")
sce.combined去批次
sce.combined = runUMAP(sce.combined, dimred = "PCA")
colnames(reducedDim(sce.combined, "UMAP")) = c("UMAP1", "UMAP2")
ggplot(data.frame(reducedDim(sce.combined, "UMAP"))) +
geom_point(alpha=0.3,aes(x = UMAP1, y = UMAP2, color = factor(sce.combined$sample_id)),size=0.7) +
labs(color = "Sample") +
theme_classic() +
scale_color_manual(breaks = c("Sec1_Anterior", "Sec1_Posterior", "Sec2_Anterior","Sec2_Posterior"),
values=c("steelblue1", "peru", "green","mediumpurple"))+
theme(axis.line = element_line(colour = 'black', size = 1.5))+
coord_fixed()harmony
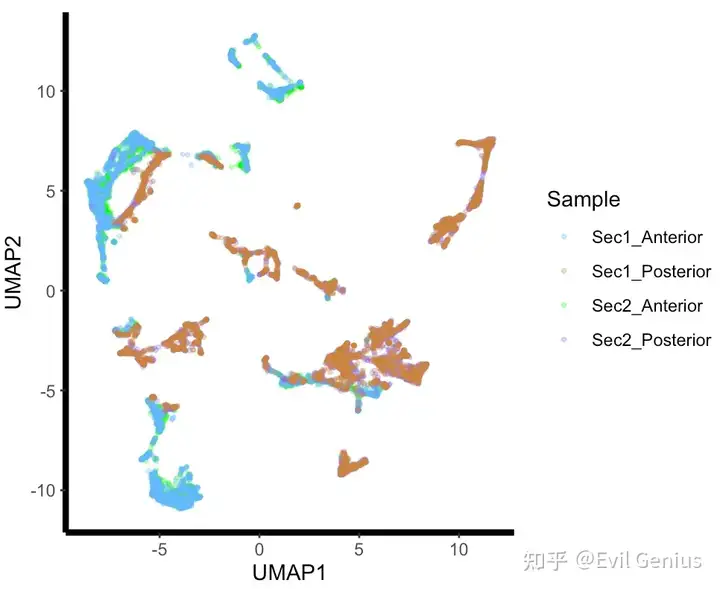
colData(sce.combined)$sample_id<-as.factor(colData(sce.combined)$sample_id)
sce.combined = RunHarmony(sce.combined, c("sample_id"), verbose = T)
sce.combined = runUMAP(sce.combined, dimred = "HARMONY", name = "UMAP.HARMONY")
colnames(reducedDim(sce.combined, "UMAP.HARMONY")) = c("UMAP1", "UMAP2")
ggplot(data.frame(reducedDim(sce.combined, "UMAP.HARMONY"))) +
geom_point(alpha=0.3,aes(x = UMAP1, y = UMAP2, color = factor(sce.combined$sample_id)),size=0.7) +
labs(color = "Sample") +
theme_classic()+
scale_color_manual(breaks = c("Sec1_Anterior", "Sec1_Posterior", "Sec2_Anterior","Sec2_Posterior"),
values=c("steelblue1", "peru", "green","mediumpurple"))+
theme(axis.line = element_line(colour = 'black', size = 1.5))+
coord_fixed()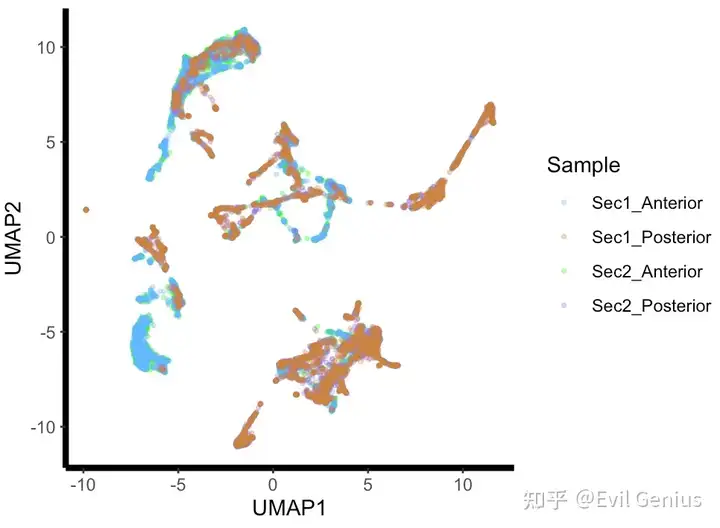
BatchCorrection
KNN Graph & Louvain Clustering
harmony<-data.frame(reducedDim(sce.combined, "HARMONY"))
harmony_umap<-data.frame(reducedDim(sce.combined, "UMAP.HARMONY"))
k <- 50
tempcom <- MERINGUE::getClusters(harmony, k, weight=TRUE, method = igraph::cluster_louvain)
dat <- data.frame("emb1" = harmony_umap$UMAP1,
"emb2" = harmony_umap$UMAP2,
"Cluster" = tempCom)
plt <- ggplot2::ggplot(data = dat) +
ggplot2::geom_point(alpha=0.4,ggplot2::aes(x = emb1, y = emb2,
color = Cluster), size = 0.9) +
ggplot2::scale_color_manual(values = rainbow(n = length(levels(tempCom)))) +
ggplot2::labs(title = "",
x = "UMAP1",
y = "UMAP2") +
ggplot2::theme_classic() +
ggplot2::theme(axis.text.x = ggplot2::element_text(color = "black"),
axis.text.y = ggplot2::element_text(color = "black"),
axis.title.y = ggplot2::element_text(),
axis.title.x = ggplot2::element_text(),
axis.ticks.x = ggplot2::element_blank(),
plot.title = ggplot2::element_text(size=15),
legend.text = ggplot2::element_text( colour = "black"),
legend.title = ggplot2::element_text( colour = "black", angle = 0, hjust = 0.5),
panel.background = ggplot2::element_blank(),
plot.background = ggplot2::element_blank(),
panel.grid.major.y = ggplot2::element_blank(),
axis.line = ggplot2::element_line(size = 1.5, colour = "black")
# legend.position="none"
) +
ggplot2::coord_fixed()
plt
Spots Transcriptional Graph
sample_ID_Sec2Ant<-matrix(0,nrow(colData(sec2_anterior)),1)
for(i in 1:nrow(colData(sec2_anterior))){
sample_ID_Sec2Ant[i,1]<-c("Sec2_Anterior")
}
sample_ID_Sec1Ant<-matrix(0,nrow(colData(sec1_anterior)),1)
for(i in 1:nrow(colData(sec1_anterior))){
sample_ID_Sec1Ant[i,1]<-c("Sec1_Anterior")
}
sample_ID_Sec2Post<-matrix(0,nrow(colData(sec2_posterior)),1)
for(i in 1:nrow(colData(sec2_posterior))){
sample_ID_Sec2Post[i,1]<-c("Sec2_Posterior")
}
sample_ID_Sec1Post<-matrix(0,nrow(colData(sec1_posterior)),1)
for(i in 1:nrow(colData(sec1_posterior))){
sample_ID_Sec1Post[i,1]<-c("Sec1_Posterior")
}
sample_ID<-rbind(sample_ID_Sec2Ant,sample_ID_Sec1Ant,sample_ID_Sec2Post,sample_ID_Sec1Post)
meta<-cbind(tempcom,sample_ID)
transcrip_edge_num <- 0
transcrip_adjacency <- list("Node1","Node2")
for(i in 1:(nrow(meta)-1)){
for(j in (i+1):nrow(meta)){
if((meta[i,1]==meta[j,1]) & (meta[i,2]!=meta[j,2])){
transcrip_edge_num <- transcrip_edge_num + 1
transcrip_adjacency$Node1[transcrip_edge_num]<-i-1
transcrip_adjacency$Node2[transcrip_edge_num]<-j-1
}
}
}
final_transcrip_adjacency<-cbind(transcrip_adjacency$Node1,transcrip_adjacency$Node2)Spots Spatial Graph
from bayestme import data
import pandas
import numpy
from bayestme import utils
temp_pos = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/tot_pos.csv',header=0,index_col=0,sep=",")
pos = temp_pos.to_numpy()
neighbors = utils.get_edges(pos=pos, layout=1)
pandas.DataFrame(neighbors).to_csv("~/Spatial/res_mouseBrain_total/spatial_neighbors.csv",index=True)Spots Similarity Graph
transcrip_neighbors<-read.csv(file = "~/Spatial/res_mouseBrain_total/transcrip_neighbors.csv")
spatial_neighbors<-read.csv(file = "~/Spatial/res_mouseBrain_total/spatial_neighbors.csv",row.names = 1)
colnames(transcrip_neighbors)<-colnames(spatial_neighbors)
similarity_neighbors<-rbind(transcrip_neighbors,spatial_neighbors)
write.csv(similarity_neighbors,file="~/Spatial/res_mouseBrain_total/similarity_neighbors.csv",quote = F,row.names = T)Bayesian Deconvolution Analysis
import pandas
import numpy
temp_counts = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/tot_ST.csv',header=0,index_col=0,sep=",")
counts = temp_counts.to_numpy()
temp_pos = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/tot_pos.csv',header=0,index_col=0,sep=",")
pos = temp_pos.to_numpy()
temp_genes = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/Gene_names.csv',header=0,sep=",")
genes = temp_genes.to_numpy()
genes = genes.reshape(-1)
temp_mask = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/tissue_mask.csv',header=0,sep=",")
mask = temp_mask.to_numpy()
mask = mask.reshape(-1)
temp_barcodes = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/Barcodes.csv',header=0,sep=",")
barcodes = temp_barcodes.to_numpy()
barcodes = barcodes.reshape(-1)
neighbors_tot = pandas.read_csv('~/Spatial/Data/MouseBrain/Total/similarity_neighbors.csv',header=0,index_col=0)
neighbors_tot.reset_index(drop=True, inplace=True)
neigh = neighbors_tot.to_numpy()
stdata= data.SpatialExpressionDataset.from_arrays(raw_counts=counts,
positions=pos,
tissue_mask=mask,
gene_names=genes,
layout=data.Layout.HEX,
barcodes=barcodes)
#len(stdata.gene_names)
stdata_stddev_filtered = gene_filtering.select_top_genes_by_standard_deviation(
stdata, n_gene=1000)
#len(stdata_stddev_filtered.gene_names)
spot_threshold_filtered = gene_filtering.filter_genes_by_spot_threshold(
stdata_stddev_filtered, spot_threshold=0.95)
stdata_filtered = gene_filtering.filter_ribosome_genes(spot_threshold_filtered)
print('{}/{} genes selected'.format(len(stdata_filtered.gene_names), len(stdata.gene_names)))
from pathlib import Path
best_lambda = 1000
best_n_components = 11
deconvolution_result = deconvolution.deconvolve(
reads=stdata_filtered.reads,
edges=stdata_filtered.edges,
n_gene=1000,
n_components=best_n_components,
lam2=best_lambda,
n_samples=100,
n_burnin=1000, # 2000 in publication
n_thin=5, # 5 in publication
bkg=False,
lda=False,
similarity_graph=neigh)
Path("./deconvolution_plots_transcrip").mkdir(exist_ok=True)
Path("./deconvolution_res_transcrip").mkdir(exist_ok=True)
deconvolution.add_deconvolution_results_to_dataset(stdata=stdata_filtered, result=deconvolution_result)
deconvolution_result.save('./deconvolution_res_transcrip/deconvolve_result.h5')
deconvolution.plot_cell_num_scatterpie(
stdata= stdata_filtered,
output_path= './deconvolution_plots_transcrip/Scatter_piechart.png')
deconvolution.plot_cell_prob(
stdata=stdata_filtered,
# result=deconvolution_result,
output_dir='./deconvolution_plots_transcrip',
output_format= 'png',
)
deconvolution.plot_cell_num(
stdata=stdata_filtered,
# result=deconvolution_result,
output_dir='./deconvolution_plots_transcrip',
output_format= 'png',
)
参考文章MUSTANG: Multi-sample spatial transcriptomics data analysis with cross-sample transcriptional similarity guidance(Patterns)
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读