ATAC-seq分析:数据介绍(2)
原创ATAC-seq分析:数据介绍(2)
原创
1. 简介
ATACseq (Assay for Transposase-Accessible Chromatin using sequencing) 使用转座酶在测序前有效地片段化可访问的 DNA(DNA可极性)。结果提供了一种绘制可访问/开放染色质基因组范围的方法。
与其他技术相比,ATACseq 有几个优点,包括:
- 所需输入材料少(> 10,000 个细胞)
- 实验所需时间短(约 4 小时)
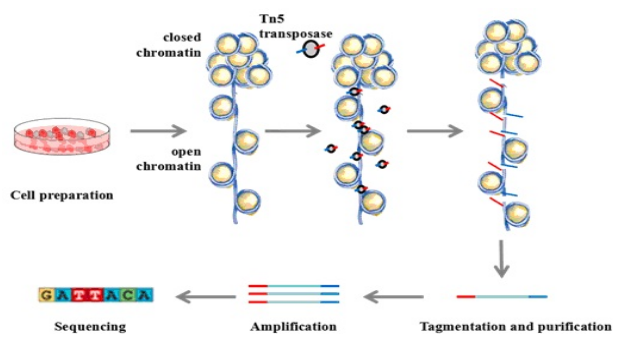
2. 酶
- 下面介绍几种不同酶获取数据的差异
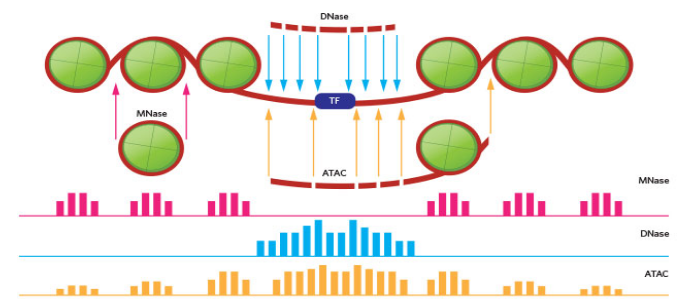
DNaseseq- 酶消化以从转录因子结合位点周围的开放染色质中提取信号。MNaseseq- 酶消化以提取代表核小体定位的信号。ATACseq- 使用转座酶并提供一种同时从单个样本的转录因子结合位点和核小体位置提取信号的方法。
3. Work
在本教程中,我们将使用一些公开的数据来了解 R 中 ATACseq 处理的一些基础知识。
将研究 ATACseq 数据在 TSS 上的比对、比对后处理和绘图。
4. 数据
本教程中,我们将使用三组已发布的数据。
4.1. data_1
第一个数据集来自原始 ATACseq 论文。我们将使用 ATACseq_50k_Rep2 示例 GEO - GSM1155958 可以从 ENA 以 FASTQ 格式获取数据。
- SAMN02192806 - here
4.2. data_2
对于第二个数据集,我们将 UCSD 的 Bing Ren 生成的 ATACseq 作为 ENCODE 联盟的一部分。它包括来自小鼠几种组织的样本。数据和示例信息的链接包含在下面的列表中。
- Liver day 12 - ENCSR302LIV
- Kidney day 15 - ENCSR023QZX
- Hindbrain day 12 - ENCSR088UYE
4.3. data_3
最后,我完全按照本次教程中的描述处理了来自 MSKCC 的 Christina Leslie 实验室的一些数据,因此我们可以在练习中回顾 ATACseq 数据的一些特征以及 ENCODE 管道处理的相同数据。
原始数据和处理后的 BAM 文件可从 ENCODEs 门户网站获得
- T-Reg - ENCSR724UJS
FQ 文件可以在此处找到 read1 和此处的 read2。我们还将使用对齐数据作为BAM 文件,该文件可在此处找到。
5. 参考数据
对于 ATACseq 分析,我们需要一些参考数据。
fasta格式的参考基因组——我们将从BSGenome Bioconductor注释包中检索。- 基因模型——我们将从
TxDb Bioconductor注释包中检索这些模型。 - Blacklists 特定于基因组的区域。这些可以在此处的 ENCODE 门户中找到
6. 已处理数据
我们从以下链接中的公共测序数据开始,并使用 Bioconductor 中的参考数据。由于其中一些处理步骤可能需要一点时间,因此我提供了指向预处理结果的链接。
来自我们对齐/排序/索引的 BAM 文件和 BAI 索引:
- SAMN02192806 - Greenleaf BAM -
Greenleaf示例的完整BAM文件在我们的Rsubread对齐、排序和索引中生成。 - SAMN02192806 - Greenleaf BAI index -
Greenleaf示例中BAM的BAI索引文件在我们的对齐、排序和索引中生成如下。
小型 BAM、peak calls 和目录结构。
- ATAC_Workshop_Essential.zip - 需要额外的文件和目录结构。
下载上述文件并解压缩 ATAC_Workshop.zip 后,您应该将 Sorted_ATAC_50K_2.bam 和 Sorted_ATAC_50K_2.bam.bai 文件移动到 ATAC_Workshop/ATAC_Data/ATAC_BAM/ 。您还应该将 RU_ATAC_Workshop.Rmd 复制到 ATAC_Workshop/ 目录,然后打开以确保所有相对路径都是正确的。
与上述相同,但具有用于计数的 BAM 以及小型 BAM、peak calls 和目录结构。
- Bigwigs - 在 IGV 中审查的 BigWigs.
- ATAC_Workshop.zip - 附加文件和目录结构。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。