? TCseq | 它可以做的不仅仅是RNA-seq的时间趋势分析!~(一文拿捏!~)
? TCseq | 它可以做的不仅仅是RNA-seq的时间趋势分析!~(一文拿捏!~)

1写在前面
又到周末了,一周开4天刀真的累啊!~?
最近也是真的很忙,根本没有时间好好地整理一下这些东西,翻一下以前写的东西分享给大家吧。?
今天介绍的是TCseq包,可以用于RNA-seq, ATAC-seq, ChIP-seq。?
与RNA-seq不同,ATAC-seq、ChIP-seq数据的基因组感兴趣区域不是预先确定的,而是特定于每个实验条件的,限制了条件之间的后续差异分析。
今天就试试表观遗传学的time course分析吧。?
2用到的包
rm(list = ls())
library(tidyverse)
library(TCseq)
3示例数据
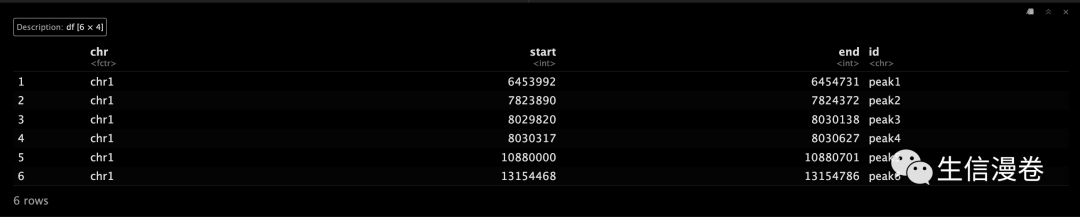
载入BED文件。?
data("genomicIntervals")
head(genomicIntervals)
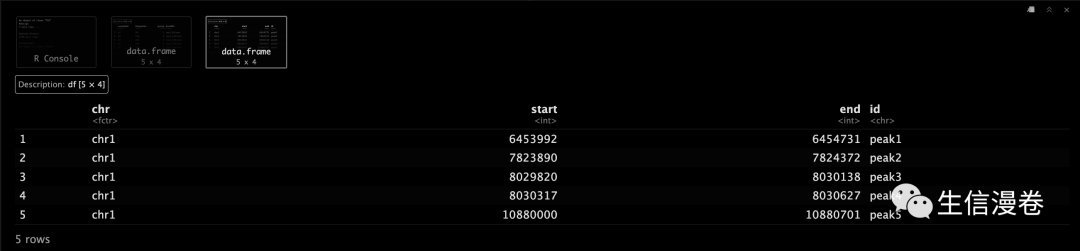
载入实验设计。?
data("experiment_BAMfile")
head(experiment_BAMfile)

4创建TCA文件
这个包自己设计了一个TCA的格式,大家来试一下吧。?
tca <- TCA(design = experiment_BAMfile, genomicFeature = genomicIntervals)
tca
有的实验设计是没有bam文件的。?
data("experiment")
data("countsTable")
tca <- TCA(design = experiment, genomicFeature = genomicIntervals, counts = countsTable)
tca

counts文件也可以后续加入到TCA文件中去。?
counts(tca) <- countsTable
还有一种就是把已有的RangedSummarizedExperiment或SummarizedExperiment 文件转为TCA文件。
安全无痛,一键完成。?
library(SummarizedExperiment)
se <- SummarizedExperiment(assays=list(counts = countsTable), colData = experiment)
tca <- TCAFromSummarizedExperiment(se = se, genomicFeature = genomicIntervals)
5差异分析
这里是基于edgeR实现的。?
小试牛刀!~??
tca <- DBanalysis(tca)
当然你也可以过滤一下表达比较低的。?
这里的过滤条件是仅保留具有两个或更多样本read counts超过10的基因组区域。??
tca <- DBanalysis(tca, filter.type = "raw", filter.value = 10, samplePassfilter = 2)
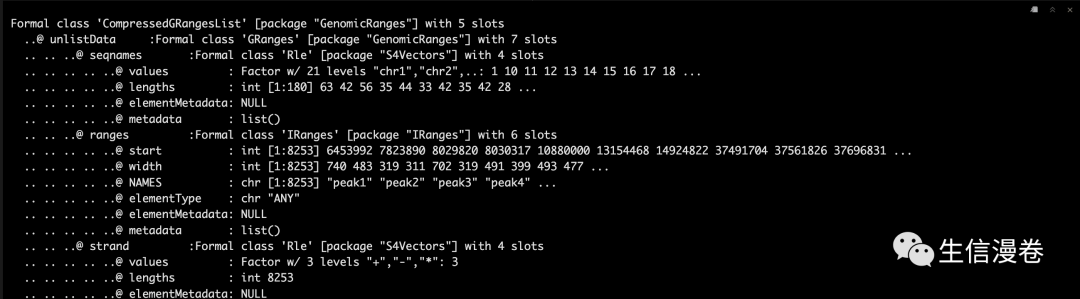
接着我们获取一下指定时间点之间的差异分析结果:?
DBres <- DBresult(tca, group1 = "0h", group2 = c("24h","40h","72h"))
str(DBres, strict.width = "cut")
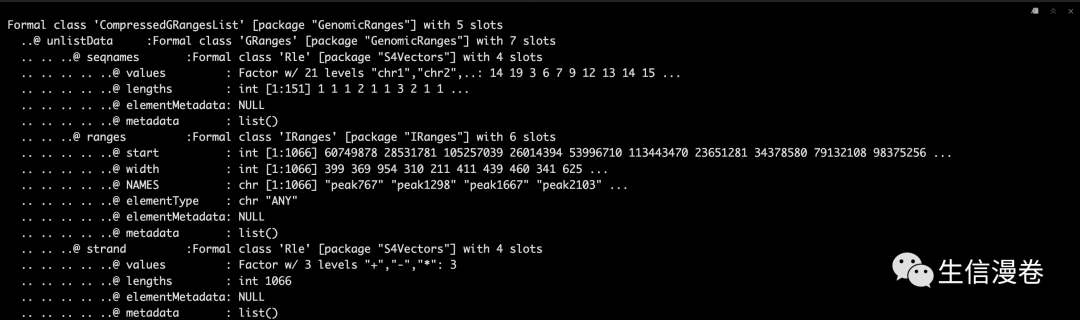
再选取一下有意义的,这里的默认条件是log2-fold > 2 or log2-fold < -2, adjusted p-value < 0.05。
DBres.sig <- DBresult(tca, group1 = "0h", group2 = c("24h","40h","72h"), top.sig = TRUE)
str(DBres.sig, strict.width = "cut")
6时间模式分析
6.1 创建相关文件
先建个timecourseTable。?
这里value可以是counts也可以是logFC?
1?? logFC
# values are logFC
tca <- timecourseTable(tca, value = "FC", control.group = "0h", norm.method = "rpkm", filter = TRUE)
2?? counts
# values are normalized read counts
tca <- timecourseTable(tca, value = "expression", norm.method = "rpkm", filter = TRUE)
前面的filter设置会顾虑掉任意两个时间点之间没有显着变化的基因区域。?
tcTable可以查看一下结果。?
t <- tcTable(tca)
head(t)

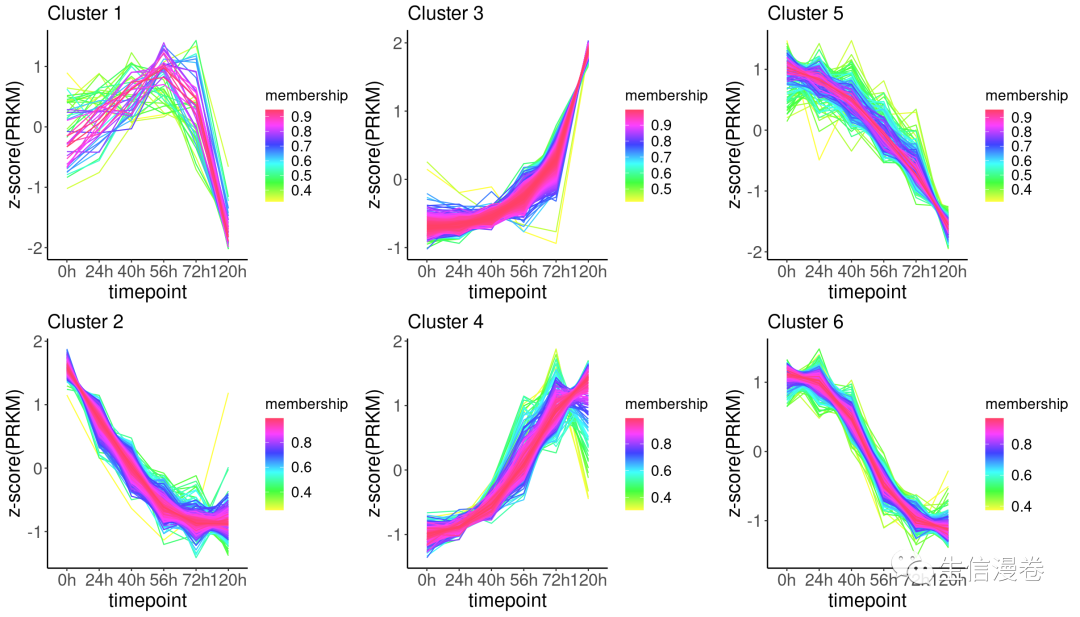
6.2 聚类分析
聚类方式有几种可供大家选择:?
"km" (kmeans); "pam" (partitioning around medoids); "hc" (hierachical clustering); "cm" (cmeans)'
tca <- timeclust(tca, algo = "cm", k = 6, standardize = TRUE)
6.3 可视化
p <- timeclustplot(tca, value = "z-score(PRKM)", cols = 3)
7小作业
大家试试如何根据membership挑选各个cluster最重要的基因区域。?

最后祝大家早日不卷!~