空间转录组niche通路富集
原创空间转录组niche通路富集
原创
追风少年i
发布于 2024-04-07 11:08:25
发布于 2024-04-07 11:08:25
通路富集;2、python版本的“hotspot”
首先是第一个内容,我们需要拿到如下结果:
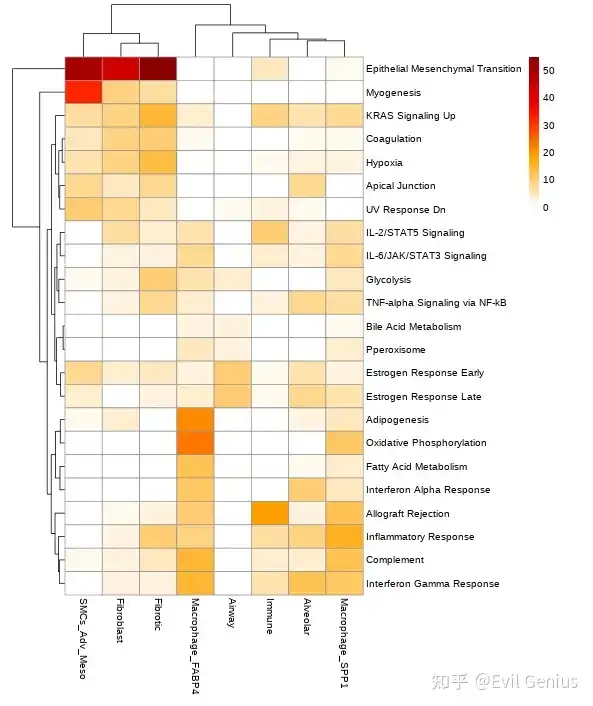
关于生态位的通路富集,之前分享过R版本,内容在
10X空间转录组数据分析之细胞类型与生物学通路的空间依赖性
空转第10课共定位内容补充(通路 && 细胞类型)
会拿到如下结果
所以大家要注意,空间类的通路富集是需要考虑空间位置关系的。
这些内容都在空转系列课程上讲过了,感兴趣的大家可以回看。
我们开始python版本的生态位通路富集,大家尽量都把脚本封装好,运行的时候就方便的多,测试的时候需要一步一步来。
import scanpy as sc
import squidpy as sq
import numpy as np
import pandas as pd
import os
import sys
import seaborn as sb
import matplotlib.pyplot as plt
from matplotlib import colors
#import scvi
import anndata as ad
import warnings
warnings.filterwarnings("ignore")
from collections import Counter
import ipywidgets as widgets
from ipywidgets import interact, interact_manual
plt.rcParams['figure.figsize'] = (6, 6)
from IPython.core.display import display, HTML
import random
#Define a colour map for gene expression
colors2 = plt.cm.Reds(np.linspace(0, 1, 128))
colors3 = plt.cm.Greys_r(np.linspace(0.7,0.8,20))
#colorsComb = np.vstack([colors3, colors2])
#mymap = colors.LinearSegmentedColormap.from_list('my_colormap', colorsComb)
from matplotlib import colors
colorsComb = np.vstack([plt.cm.Reds(np.linspace(0, 1, 128)), plt.cm.Greys_r(np.linspace(0.7, 0.8, 0))])
mymap = colors.LinearSegmentedColormap.from_list('my_colormap', colorsComb)
# Helper function to split list in chunks
def chunks(lista, n):
for i in range(0, len(lista), n):
yield lista[i:i + n]
plt.rcParams['figure.figsize'] = (6, 5)
sc.set_figure_params(dpi=100, vector_friendly=True)
def mysize(w, h, d):
fig, ax = plt.subplots(figsize = (w, h), dpi = d)
return(fig.gca())
plt.rcParams['figure.figsize'] = (6, 5)
sc.set_figure_params(dpi=100, vector_friendly=True)
sc.settings.figdir = "./figures/"
## frequently used variables
from matplotlib import colors
import matplotlib.pyplot as plt
colorsComb = np.vstack([plt.cm.Reds(np.linspace(0, 1, 128)), plt.cm.Greys_r(np.linspace(0.7, 0.8, 0))])
mymap = colors.LinearSegmentedColormap.from_list('my_colormap', colorsComb)
## Along these Lines, a colourmap diverging from gray to red
gray_red = colors.LinearSegmentedColormap.from_list("grouping", ["lightgray", "red", "darkred"], N = 128)
## Some more Colour Maps
gray_violet = colors.LinearSegmentedColormap.from_list("grouping", ["lightgray", "mediumvioletred", "indigo"], N = 128)
gray_blue = colors.LinearSegmentedColormap.from_list("grouping", ["lightgray", "cornflowerblue", "darkblue"], N = 128)
def mysize(w, h, d):
fig, ax = plt.subplots(figsize = (w, h), dpi = d)
return(fig.gca())
#plt.rcParams['figure.figsize'] = (6, 5)
#sc.set_figure_params(dpi=120, vector_friendly=True)
import matplotlib.colors as colors
c_low = colors.colorConverter.to_rgba('orange', alpha = 0)
c_high = colors.colorConverter.to_rgba('red',alpha = 1)
cmap_transparent = colors.LinearSegmentedColormap.from_list('rb_cmap',[c_low, c_high], 512)
import matplotlib.colors as colors
c_low2 = colors.colorConverter.to_rgba('green', alpha = 0)
c_high2 = colors.colorConverter.to_rgba('darkblue',alpha = 1)
cmap_transparent2 = colors.LinearSegmentedColormap.from_list('rb_cmap',[c_low2, c_high2], 512)加载数据,注意这里要完成单细胞空间的联合分析进行空间注释,这个内容在课程上也都讲过了,不再重复了。
##load
adata_vis = sc.read(f".h5ad")
####细胞类型,根据自己的项目选择
cts = ['AT0', 'AT1', 'AT2', 'Aberrant basaloid', 'Adventitial fibroblast', 'Alveolar fibroblast',
'Artery', 'B/Plasma', 'Basal', 'Basophil/Mast', 'Bronchial Vessel', 'Capillary', 'Capillary Aerocyte',
'Ciliated', 'Ciliated SFTPB+/SCGB1A1+', 'Dendritic', 'Ionocyte', 'Lymphatic', 'Macrophage C1Q hi',
'Macrophage CHI3L1+/CD9 hi/', 'Macrophage FABP4+', 'Macrophage IL1B+', 'Macrophage LYVE1+',
'Macrophage RETN+/VCAN+', 'Mesothelial', 'Monocyte', 'Mucous', 'Myofibroblast', 'NK',
'Peribronchial fibroblast', 'Pericyte', 'Smooth Muscle', 'T cell', 'TB-SC', 'Vein', 'pDC', 'preTB-SC/RAS']生态位marker的计算
## All Markers
sc.tl.rank_genes_groups(adata_vis, groupby = 'Niche_NMF', groups = "all", use_raw = False, method = "wilcoxon", pts= True,)
sc.pl.rank_genes_groups(adata_vis)
## Combine into one Data Frame (comparable to Marker Table)
result = adata_vis.uns['rank_genes_groups']
allMarkers = []
for cluster in result['names'].dtype.names:
current = pd.DataFrame({"gene": result["names"][cluster], "score": result["scores"][cluster],
"logfoldchange": result["logfoldchanges"][cluster], "pval": result["pvals"][cluster],
"pval_adj": result["pvals_adj"][cluster],
"pct_within":result["pts"].loc[result["names"][cluster]][cluster],
"pct_outside":result["pts_rest"].loc[result["names"][cluster]][cluster], "cluster": cluster})
allMarkers.append(current)
allMarkers = pd.concat(allMarkers)
allMarkers.head()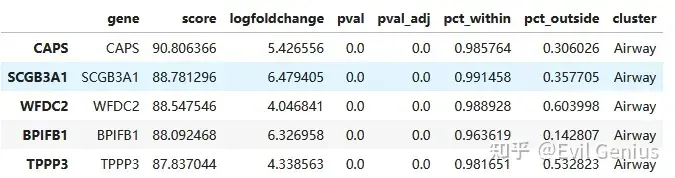
## Write to file
#folder =
allMarkers.to_csv("AllMarkers_human_Niche_NMF_CM.txt", sep = "\t", index = False)
allMarkers.drop("pval", axis=1, inplace=True)
for i in adata_vis.obs.Niche_NMF.cat.categories:
allMarkers_subset =allMarkers[allMarkers["cluster"]== i ]
allMarkers_subset.to_csv("AllMarkers_human_Niche_NMF_%s.txt" %i, sep = "\t", index = False)
marker3 = allMarkers
marker3 = marker3[marker3['pct_outside'] < 0.25]
marker3 = marker3[marker3['pct_within'] > 0.25]
marker3 = marker3[marker3['logfoldchange'] > 1]
marker3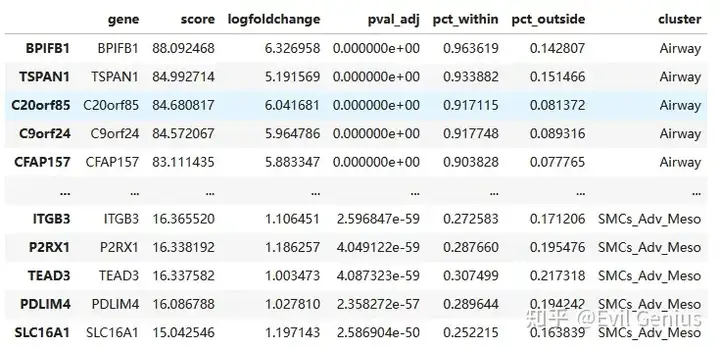
绘图marker
#Code zum filtern hast ja schon, die folgenden Zeilen w?hlen dann die top n gene per cluster aus (im gleichen ordering wie das dendrogram)
marker3.sort_values(by = ["cluster", "logfoldchange"], ascending = [True, False], inplace = True)
#Get ordering as in Dendrogram
sc.tl.dendrogram(adata_vis, groupby = "Niche_NMF")
order = adata_vis.uns["dendrogram_Niche_NMF"]["dendrogram_info"]["ivl"]
#Get top genes from Markers Table
n_genes = 10
genes = []
for typ in order:
curgenes = marker3.loc[marker3.cluster == typ, "gene"].values[0:n_genes]
genes = genes + [g for g in curgenes if g not in genes]
sc.pl.matrixplot(adata_vis, var_names = genes, standard_scale = "var", groupby = "Niche_NMF", dendrogram = True,figsize=(20,3),layer="log1p",
save = "Allmarkers_matrixplottop%s_per_Niche_NMF_CM_025background_025celltype10logfc.pdf" %n_genes)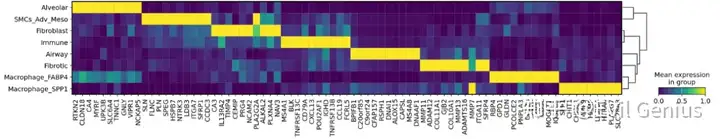
生态位通路富集
##gene set enrichment on DGe genes between Niche_NMF_CM
allMarkers =allMarkers[allMarkers["pval_adj"]<0.05]
import gseapy
#Available databases : ‘Human’, ‘Mouse’, ‘Yeast’, ‘Fly’, ‘Fish’, ‘Worm’
gene_set_names = gseapy.get_library_name(organism='Human')
allMarkers_subset =allMarkers[allMarkers["cluster"]== "Alveolar_AT1"]
allMarkers_subset =allMarkers_subset[allMarkers_subset["pval_adj"] < 0.05 ]
allMarkers_subset =allMarkers_subset[allMarkers_subset["logfoldchange"] > 0.5 ]
#?gseapy.enrichr
for i in adata_vis.obs.Niche_NMF.cat.categories:
genelist = sc.get.rank_genes_groups_df(adata_vis, group=i,
log2fc_min=0.5,
pval_cutoff=0.05)['names'].squeeze().str.strip().tolist()
enr_res = gseapy.enrichr(gene_list=genelist,
organism='Human',
background=adata_vis.var_names,
gene_sets='GO_Biological_Process_2021',
cutoff = 0.05,)
enr_res.results["Niche"] = "%s" %i
enr_res.results= enr_res.results[enr_res.results["Adjusted P-value"] <0.05]
#enr_res.results= enr_res.results.nsmallest(n=6, columns=['Adjusted P-value'],keep="all")
enr_res.results.to_csv("./GO_enrichment/GO_enrichment_%s.txt" %i, sep = "\t", index = False)
gsea_res = pd.concat([pd.read_csv("./GO_enrichment/GO_enrichment_%s.txt" %i,sep = "\t")for i in adata_vis.obs.Niche_NMF.cat.categories], axis = 0)
gsea_res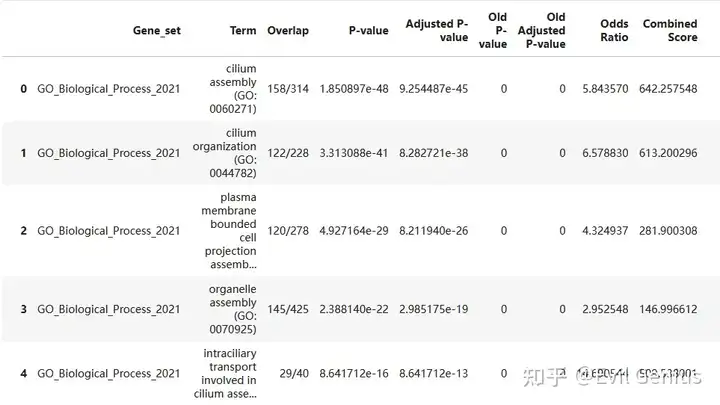
gsea_table_new = gsea_res[gsea_res["Term"].isin(gsea_table["Term"])]
gsea_table_new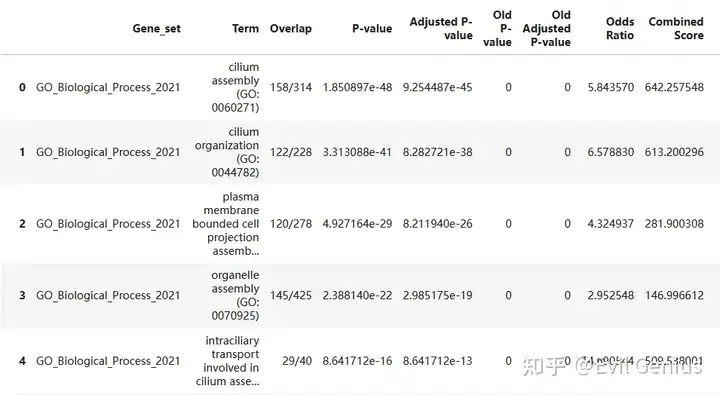
gsea_table_new = gsea_table_new.drop(["Gene_set","Overlap","P-value","Old P-value","Old Adjusted P-value","Odds Ratio","Genes","Combined Score"], axis=1)
gsea_table_new
gsea_table_new = gsea_table_new[gsea_table_new["Niche"] != "no tissue"]
gsea_table_new
gsea_table_new["Adjusted P-value"] = np.log10(gsea_table_new["Adjusted P-value"])
gsea_table_new
gsea_table_new["Adjusted P-value"] = gsea_table_new["Adjusted P-value"]*(-1)
gsea_table_new
gsea_table_new['Adjusted P-value'].values[gsea_table_new['Adjusted P-value'] > 50] = 50
gsea_table_new.sort_values(by=["Adjusted P-value"])
gsea_table_new
gsea_table_new_cm= pd.pivot(gsea_table_new,index="Term", columns="Niche", values="Adjusted P-value")
gsea_table_new_cm = gsea_table_new_cm.fillna(0)
gsea_table_new_cm
绘图
%%R -w 550 -h 700 -i gsea_table_new_cm
my_col <- colorRampPalette(c("white","orange", "red","darkred"))(50)
my_col[0] <- "white"
p<-pheatmap(gsea_table_new_cm, color= my_col)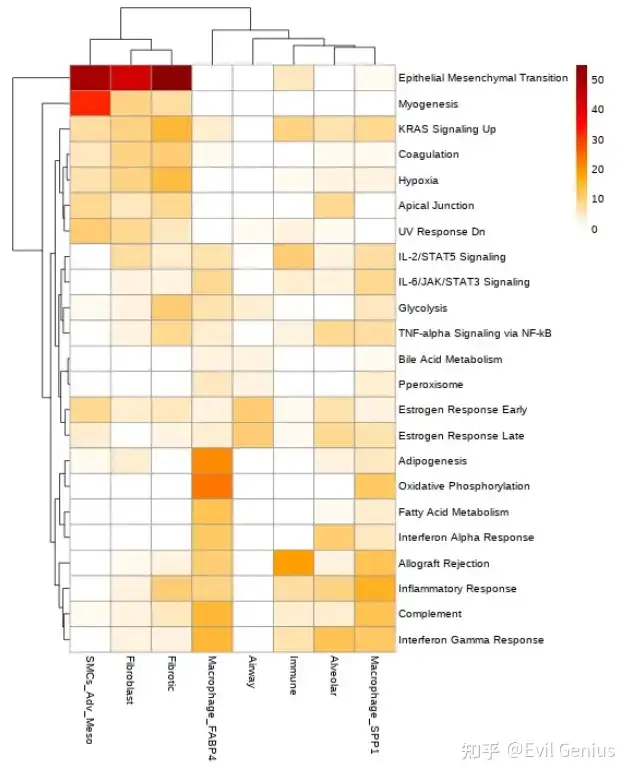
关于python版本的“hotspot”,参考文章空间转录组niche通路富集与空间“hotspot”
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录