空间邻域分析导论(CellNeighborEX)
原创空间邻域分析导论(CellNeighborEX)
原创
作者,Evil Genius
我们来做一篇导论,关于空间邻域
空间邻域分析包括分子邻域和细胞邻域,分子邻域主要研究邻域通讯,细胞邻域主要研究生态位,我已经分享了很多了,做一篇导论给大家指引一下分析方向,同时介绍邻域分析的软件CellNeighborEX。文献在CellNeighborEX: deciphering neighbor‐dependent gene expression from spatial transcriptomics data。
当然,这里主要介绍的是分子邻域,经过简单的修改就可以分析细胞邻域。
这就是周五的推文了,真正的周五,要去相亲了。
适用的空间平台,slide-seq 、华大、10X HD、 10X Visium 等。
空间细胞邻域网络图
空间邻域通讯分析大汇总
空间转录组学数据分析细胞邻域依赖的基因表达(分子邻域)
空间组学邻域分析方法更新之BANKSY
10X空间转录组之构建邻域通讯网络
空间多组学分析破译胶质母细胞瘤中的双向肿瘤-宿主相互依赖性(空间微环境)
10X空间转录组数据分析之细胞类型与生物学通路的空间依赖性
10X空间转录组数据分析之细胞的空间依赖性
好了,我们开始
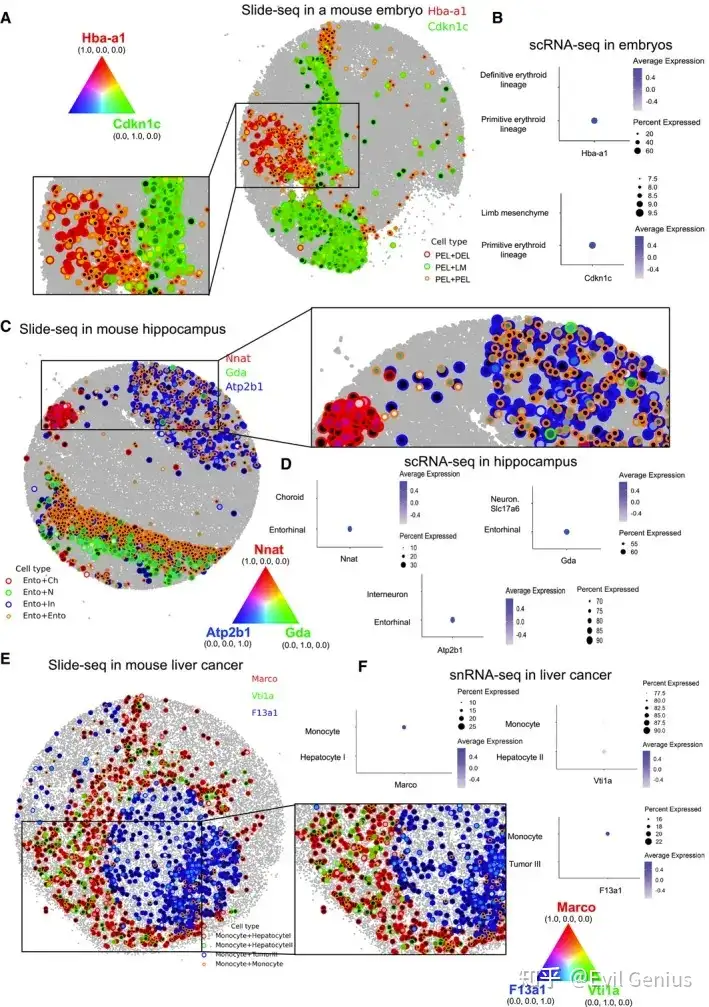
细胞已经进化出它们的通讯方法来感知它们的微环境并发送生物信号。除了使用配体和受体进行通信外,细胞还使用包括间隙连接在内的多种通道与其近邻进行通信。然而,当前的方法无法有效捕获各种微环境的影响。为了根据微环境对细胞进行分类,CellNeighborEX利用ST技术直接定位细胞或混合多个细胞的转录组。对于每种细胞类型,cellneighborx可以识别与配对细胞类型相关的不同基因集,从而提供进一步的见解。细胞根据邻近细胞的类型表达不同的基因。这些基因与关键的生物过程有关,如发育或转移。
细胞以多种方式与其微环境进行交流,包括释放可溶性分子和直接细胞接触,积极改变其转录组以响应外部信号。为了深入了解关键的生物过程,如疾病和发育,了解细胞间交流的各种方式是必不可少的。研究细胞间通讯的实验方法通常需要精心设计和复杂的设置。
最近,利用配体-受体共表达单细胞RNA-sequencing (scRNA-seq)和空间转录组学(ST)数据,可以在基因组尺度上研究细胞-细胞相互作用。使用配体-受体共表达可以推断相互作用的细胞类型对并识别细胞间信号通路,而无需依赖于复杂的实验设置。然而,它不能解释细胞直接接触所改变的单个细胞的基因表达。
越来越多的关于细胞通讯的研究表明,细胞受其微环境和邻近细胞的影响。发育中的胚胎移植实验表明,细胞直接接触可以诱导特定组织类型发育的信号。最近,物理相互作用细胞的RNA测序(PIC-seq)显示,在小鼠发育过程中,细胞根据邻近细胞的类型表达不同的基因。该研究表明,通过独立于配体-受体介导的通信的直接细胞接触,细胞具有不同的表达谱。
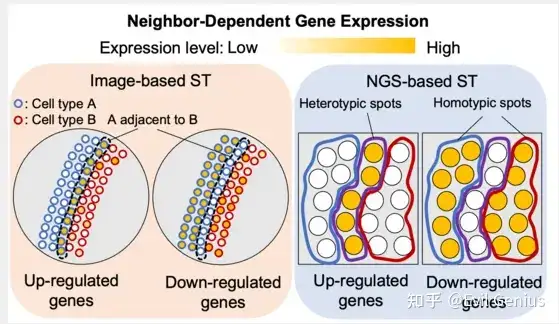
ST的最新发展为探索微环境的作用开辟了潜在的途径。空间基因表达谱使得研究细胞的转录活性以及完整组织内邻近细胞的转录活性成为可能。ST数据主要有两种类型,各有优点和局限性。基于图像的方法包括MERFISH和seqFISH使用荧光原位杂交(FISH)来可视化感兴趣的RNA信息。虽然基于图像的ST方法可以在细胞分辨率上量化RNA,但可检测的RNA种类数量仍然有限。基于下一代测序(NGS)的ST方法,如Visium和Slide-seq利用空间条形码。虽然基于NGS的方法可以公正地描绘转录组,但条形码可能与多个细胞或细胞部分的转录组混合物相关联,这取决于条形码的位置和分辨率,这使得很难检测到细胞微环境改变的基因表达。
已经开发了许多计算工具来从ST数据中理解细胞-细胞相互作用。CellphoneDB v.3.0(目前已经V5了)、MESSI、SpaTalk和stLearn使用配体-受体对的共表达来研究细胞通讯。然而,由于直接接触,配体-受体共表达不能完全捕获细胞-细胞相互作用。SVCA将基因表达变异的来源分解为内在效应、环境效应和细胞-细胞相互作用。它解释了基因表达和细胞间相互作用之间的关系。然而,SVCA不具有检测与细胞接触相关的基因表达变化的功能,并且它们的策略仅针对基于图像的ST数据进行了优化。由于MISTy量化了不同空间背景对感兴趣标记表达的信息,因此可以研究近邻对配受体表达的影响。然而,MISTy需要预先选择标记基因列表来发现潜在的相互作用,并且它还没有被设计成以一种公正的方式识别与细胞接触相关的基因表达变化。DeepLinc从ST数据重建细胞相互作用网络。将三个最近的邻居作为直接接触,DeepLinc发现了有助于细胞类型之间相互作用的特征基因,并推断出它们之间的近端相互作用。然而,它并没有揭示特征基因和相互作用的细胞类型之间的特定关系。C-SIDE检查上调和下调的基因取决于接近某种细胞类型。由于细胞类型之间的相互作用是基于细胞密度而不是细胞接触来定义的,因此C-SIDE不适用于研究细胞接触依赖性基因表达。NCEM研究了依赖于当地环境的转录组变化,但它并没有被设计用于研究细胞接触对基因表达的影响,特别是对于基于NGS的数据。即使在低分辨率的Visium数据中,NCEM也将一个Barcode视为单个细胞类型,因此它不会研究一个点内多个细胞类型直接接触的影响。虽然空间环境已被应用于研究细胞-细胞相互作用,但与细胞接触相关的转录组学变化尚未得到充分探索。无论数据类型如何,检测受细胞接触影响的基因仍然具有挑战性。
分析方法概述
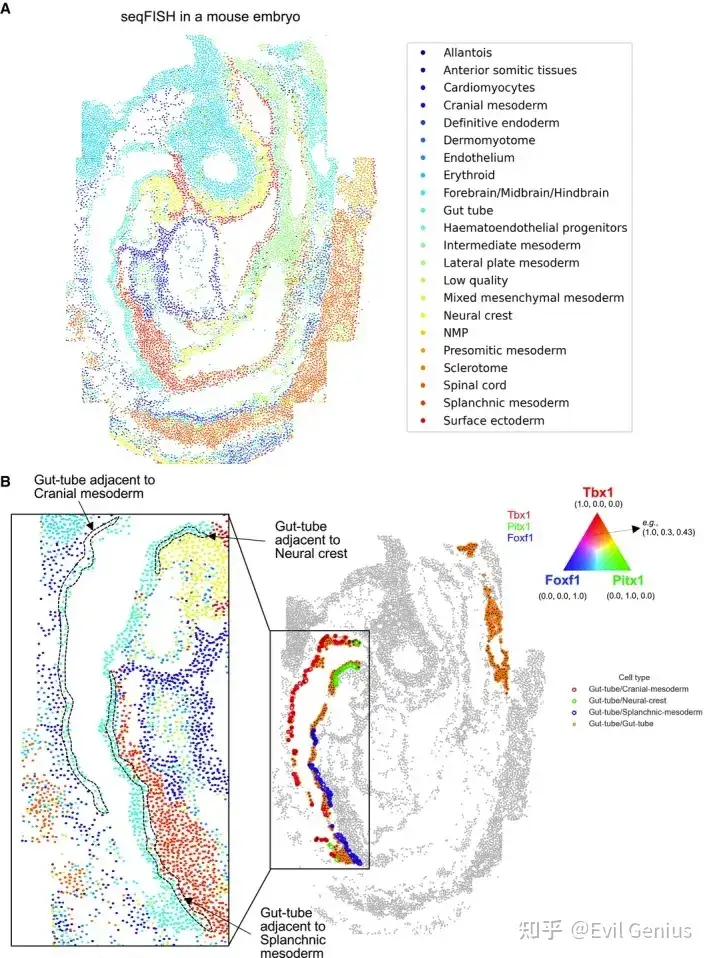
cellneighborEX对图像和基于NGS的ST数据的直接邻居定义不同。在基于图像的ST数据中,可以获得精确的细胞位置,cellneighborEX使用Delaunay三角剖分、径向距离或k近邻来找到最近的邻居。径向距离和KNN都需要先验知识。KNN对于细胞均匀分布在组织上是有用的。当知道感兴趣的细胞之间的距离时,径向距离是有用的。Delaunay三角测量可以单独使用,也可以与径向距离结合使用。
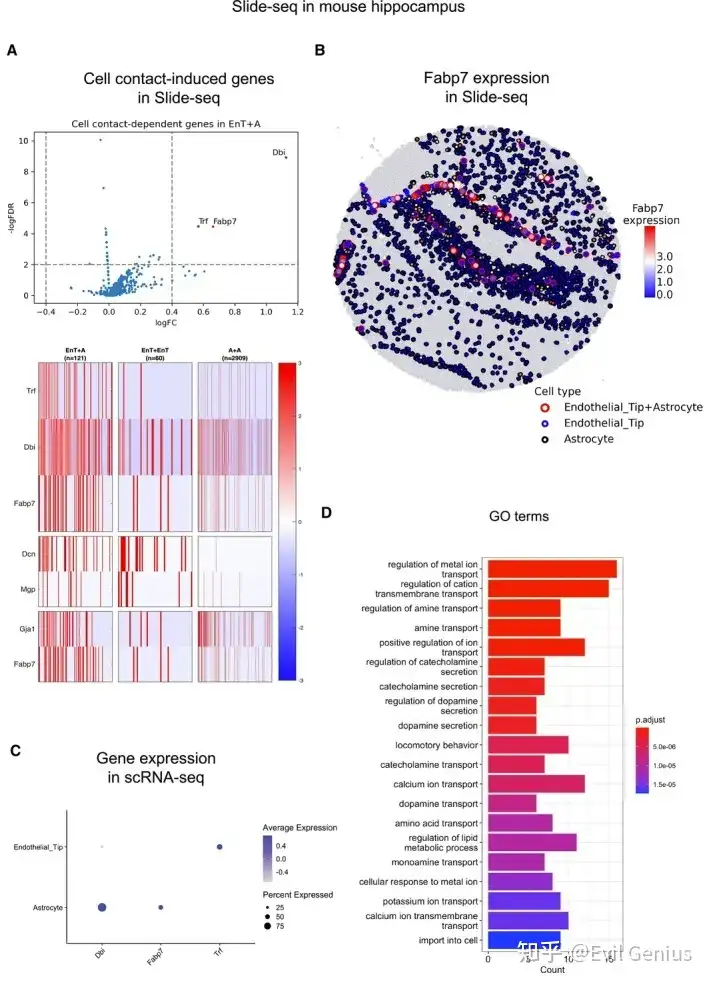
cellneighborEX根据最近邻居的细胞类型将所有细胞分为两组。同型邻居由相同的细胞类型组成。异型邻居由不同的细胞类型组成。通过比较异型邻居和同型邻居的转录组,可以衡量邻近细胞的影响(log ratio > 0.4, p值< 0.01,FDR < 0.05)。该策略也适用于基于NGS的ST方法,在细胞分割后具有高分辨率(< 1 μm分辨率)。
基于NGS的ST方法没有明确给出细胞的位置,Barcode包含来自多个细胞的混合转录组。cellneighborEX利用具有多种细胞类型的SPOT(或异型SPOT)作为细胞接触的证据。当SPOT直径接近细胞分辨率时,该策略在研究邻近依赖基因表达方面是有效的。例如,10 μm分辨率的ST方法Slide-seq具有同型SPOT(在一个SPOT中有相同的细胞类型)和异型SPOT(在一个SPOT中有两种或两种以上的细胞类型),并且大多数异型SPOT由两种细胞类型组成(97%的SPOT由一种或两种细胞类型组成)。cellneighborEX使用RCTD来分解Slide-seq数据中SPOT的细胞类型。RCTD还根据scRNA-seq数据估计ST数据中每个SPOT的细胞类型比例。将异型SPOT的转录组与同型SPOT的转录组进行比较,鉴定邻居依赖基因(对数比> 0.4,p值< 0.01)。为了进一步确认所鉴定的邻居依赖基因的统计显著性,cellneighborEX通过创建人工异型SPOT生成null model。利用RCTD估计的异型SPOT细胞型比例信息,将两个随机的同型SPOT合并形成人工异型SPOT。人工异型SPOT代表两种不同的细胞类型只是结合在一起,没有细胞间的相互作用。分析表明,邻居依赖基因对这些人工SPOT也有差异表达(FDR < 0.01)。

接下来是分析结果示例
我们来看看示例代码,给了4个示例
我们来看其中的一个
import pandas as pd
import os
import CellNeighborEX示例数据,单细胞空间联合分析的方法是RCTD
# Check the path of your root directory.
os.getcwd()
'/Users/kimh15/Downloads'
# Make a folder to save data files.
if not os.path.exists('Datasets'):
os.makedirs('Datasets')
# Download data files.
# (i) pre-processed expression data
wget https://figshare.com/ndownloader/files/42334083 -O Datasets/SSliver_log_data.txt
wget https://figshare.com/ndownloader/files/42334077 -O Datasets/SSliver_cell_id.txt
wget https://figshare.com/ndownloader/files/42334080 -O Datasets/SSliver_gene_name.txt
# (ii) data of annotated cell types and spatial coordinates
wget https://figshare.com/ndownloader/files/42333705 -O Datasets/SSliver_RCTD.csv加载数据
# Set the path of data files regarding annotated cell types.
path = '/Users/kimh15/Downloads/Datasets/'
df_processed = pd.read_csv(path + 'SSliver_RCTD.csv', header=0)
df_processed.head(5)barcode | first_type | second_type | celltype1 | celltype2 | x | y | prop1 | prop2 |
|---|---|---|---|---|---|---|---|---|
CTTGTGGTTGCAGA | 6 | 2 | TumorIII | Monocyte | 3670.6 | 2839.7 | 0.532488 | 0.149015 |
TTTTTTTTTTTTTT | 5 | 2 | HepatocyteI | Monocyte | 2584.0 | 1702.6 | 0.470880 | 0.099647 |
ACATGTCTATGTTA | 5 | 1 | HepatocyteI | HepatocyteII | 5226.2 | 4028.4 | 0.737157 | 0.261249 |
GCGCTTTCGTTCCA | 5 | 1 | HepatocyteI | HepatocyteII | 4708.4 | 1851.7 | 0.825412 | 0.167525 |
ACAATTGTCTGCAC | 4 | 4 | TumorII | TumorII | 1385.5 | 4176.6 | 0.745914 | 0.000000 |
计算每个spot的细胞丰度
# All categorzied files (index_, matchComb_, neiCombUnique_, prop_ .csv) are saved in the "categorized_data folder" in the root directory.
CellNeighborEX.categorization.generate_input_files(data_type = "NGS", df = df_processed, sample_size=30, min_sample_size=1)
# Set the path of the directory where all the categorized data files are saved.
path_categorization = '/Users/kimh15/Downloads/categorized_data/'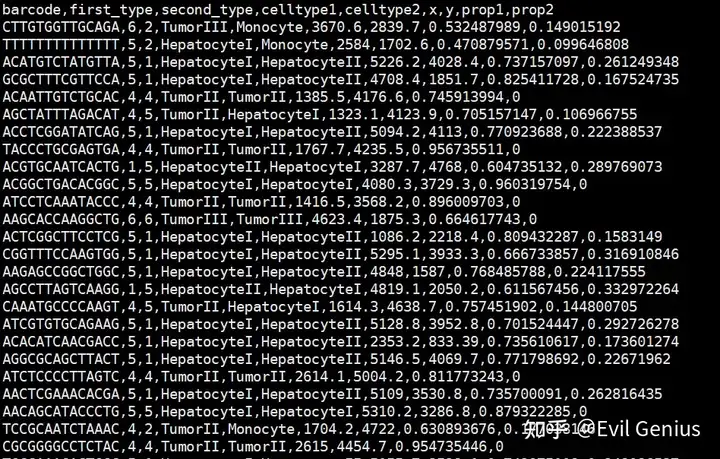
# Save the data into dataframes. 矩阵信息
####Barcode
df_cell_id = pd.read_csv(path + "SSliver_cell_id.txt", delimiter="\t", header=None)
####gene
df_gene_name = pd.read_csv(path + "SSliver_gene_name.txt", delimiter="\t", header=None)
####基因表达矩阵
df_log_data = pd.read_csv(path + "SSliver_log_data.txt", delimiter="\t", header=None)空间依赖基因的表达
# Set argument values for CellNeighborEX.DEanalysis.analyze_data().
data_type = "NGS" # Image: image-based ST data, NGS: NGS-based ST data
lrCutoff = 0.4 # log ratio
pCutoff = 0.01 # p-value
pCutoff2 = 0.01 # false discovery rate
direction = 'up' # up: up-reguated genes, down: down-regulated genes
normality_test = False # True: depending on the result of the normality test, the statistical test is determined. If the data is normal, the parametric test is used. Otherwise, the non-parametric test is used.
# False: when sample size (number of cells/spots) is larger than 30, the parameteric test is used. Otherwise, the non-parametric test is used.
top_genes = 10 # Top 10 DEGs are annotated in the volcano plot.
# If save=True, all result files (DEG list: csv, heatmaps and volcano plots: pdf, gene expression values: txt) are saved in the "DE_results" folder in the root directory.
DEG_list = CellNeighborEX.DEanalysis.analyze_data(df_cell_id, df_gene_name, df_log_data, path_categorization, data_type, lrCutoff, pCutoff, pCutoff2, direction, normality_test, top_genes, save=True)
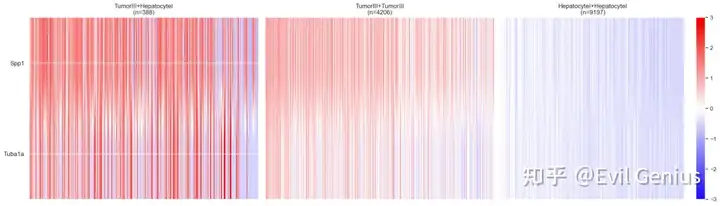
Visualize neighbor-dependent gene expression in the spatial context
# Select a cell type and a DEG for spatial visualization and then load the data.
# For example, F13a1 is one of up-regulated genes identified from the heterotypic spots of TumorIII+Monocyte.
path_selected = '/Users/kimh15/Downloads/DE_results/TumorIII+Monocyte/'
column_names = ['barcode', 'logdata', 'zscore']
heterotypic = pd.read_csv(path_selected + "TumorIII+Monocyte_F13a1.txt", delimiter=",", names = column_names)
homotypic1 = pd.read_csv(path_selected + "TumorIII+TumorIII_F13a1.txt", delimiter=",", names = column_names)
homotypic2 = pd.read_csv(path_selected + "Monocyte+Monocyte_F13a1.txt", delimiter=",", names = column_names)
heterotypic['type'] = 'TumorIII+Monocyte'
homotypic1['type'] = 'TumorIII+TumorIII'
homotypic2['type'] = 'Monocyte+Monocyte'
df_exp = pd.concat([heterotypic, homotypic1, homotypic2])
# Set parameter values.
df_bg, df_red, df_blue, df_black = CellNeighborEX.visualization.set_parameters(df_processed, df_exp, beadsize_bg=10, edgecolor_bg=(0.85,0.85,0.85), beadcolor_bg=(0.85,0.85,0.85), beadsize_red=600, beadsize_blue=200, beadsize_black=200, type_red='TumorIII+Monocyte', type_blue='TumorIII+TumorIII', type_black='Monocyte+Monocyte')
# Get the spatial map.
# zorder_red, zorder_blue, and zorder_black are parameters that determine the drawing order in the spatial map.
# If save=True, the spatial map (F13a1.pdf) is saved in the "spatialMap" folder in the root directory.
CellNeighborEX.visualization.get_spatialPlot(df_bg, df_red, df_blue, df_black, label_red='TumorIII+Monocyte', label_blue='TumorIII', label_black='Monocyte', label_gene='F13a1', zorder_red=3.0, zorder_blue=2.0, zorder_black=4.0, figsize=(28,28), save=True)
代码示例在CellNeighborEX — CellNeighborEX 0.0.5 documentation
https://cellneighborex.readthedocs.io/en/latest/index.html
大家根据自己的情况做一点修改就可以运用到自己的数据了。
生活很好,有你更好
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。