差异分析不是这样做的……

那天在Frontiers in Immunology 偶然一瞥,看到一篇新鲜出炉的纯生信文章
大喜?!纯生信文章还能往这里投?赶紧学习学习,然后……我就看到了这张神奇的图?
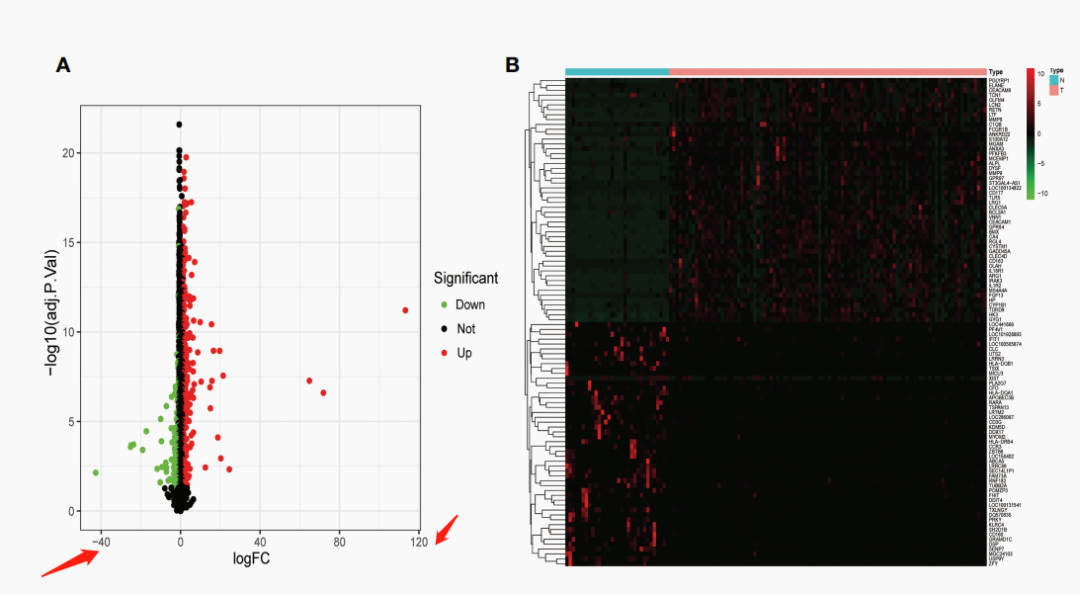
原文对差异分析是这么描述的:Using R software’s limma package , differentially expressed genes (DEGs) between septic shock cohort and control cohort were analyzed, with the following criterion: p<0.05 and |fold change (FC)| > 1.
竟然是直接用fold change来作为阈值的,一般差异分析用的更多的是log2FC,这样数字不会特别离谱。
毕竟,log2FC中的FC即 fold change,表示两个样本/组间表达量的比值,对其取以2为底的对数之后才是log2FC。
举几个例子:基因A 在肿瘤和正常组织中的表达量分别为2和4,那么比值为2,即FC=2,此时log2FC=1; 基因B 在肿瘤和正常组织中的表达量均为2,此时比值为1,即FC=1,此时log2FC=0; 基因C 在肿瘤和正常组织中的表达量分别为2和64,那么比值为32,即FC=32,此时log2FC=5。 最后夸张一下!! 基因D 在肿瘤和正常组织中的表达量分别为2和1024,那么比值为512,即FC=512,此时log2FC=9。
这样一算,你大概就能明白上面那张图问题出在哪里了吧~
但是光说不练,纸上谈兵,我们还是自己上手分析一下这个数据集,验证一下自己的猜想——
这里的上下调基因取得是top30的哈~,代码放在下面:
cg = rownames(deg)[deg$change != "stable"]
deg1 <- deg[cg,]
library(dplyr)
top_up <- deg1 %>% top_n(n=30,wt=logFC) %>% rownames()
top_down <- deg1 %>% top_n(n=-30,wt=logFC) %>% rownames()
top_all <- c(top_up,top_down)
n1 <- n[top_all,]
# 由于 ComplexHeatmap 包并不会对数据进行标准化,为了让图形更好看,我们先手动对数据进行标准化
exp <- apply(n1, 1, scale)
rownames(exp) <- colnames(n1)
exp <- t(exp)
然后是画图参数的设置:
col_fun = colorRamp2(c(-2, 0, 2), c("#2fa1dd", "white", "#f87669"))
top_annotation = HeatmapAnnotation(
cluster = anno_block(gp = gpar(fill = c("#2fa1dd", "#f87669")),
labels = c("normal","case"),
labels_gp = gpar(col = "white", fontsize = 12)))
p4 <- pheatmap(exp,name = "expression",
col = col_fun,
column_split = group_list,
top_annotation=top_annotation,
row_split = rep(c("UP","DOWN"), each = 30),
fontsize_row=5,
column_title = NULL,
show_colnames = F)
p4
p4 <- as.ggplot(p4)
最后是拼图的基操:
library(patchwork)
p <- p3+p4 ##p3就是火山图
ggsave(p,filename = "../outputs/article.pdf",width = 15,height = 9)
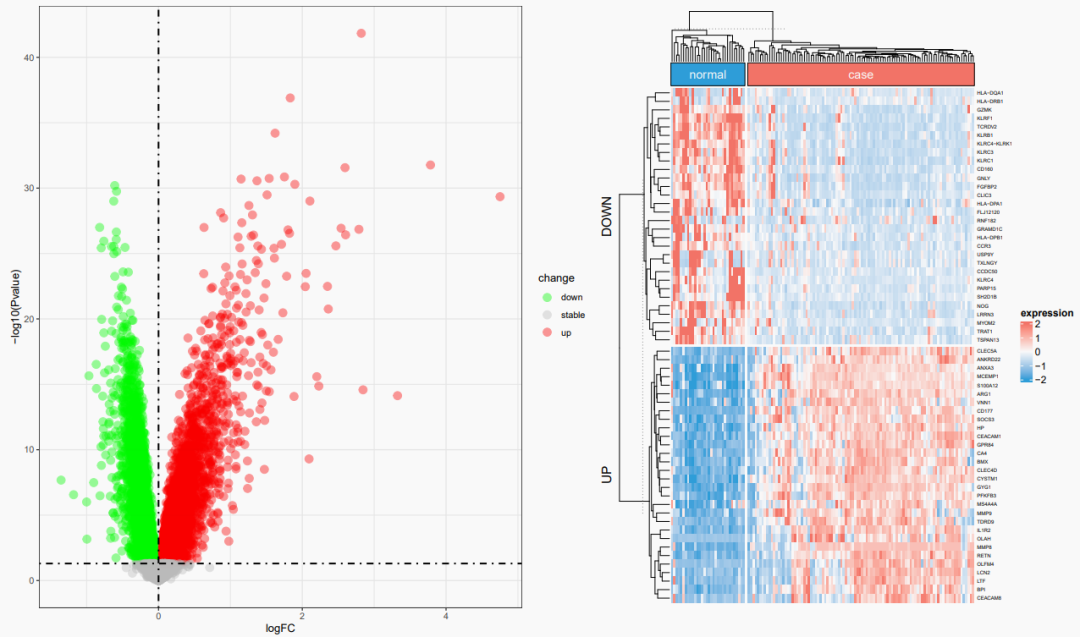
为什么我的图和文章里的图迥然不同呢?
相信大家只要对表达量矩阵有一定的熟悉,就应该知道,有的数据集下载以后,需要先观察探针在每一个样本中的表达量数据,一般数值不大于20的话,说明这个矩阵已经是被取过log的,否则的话是需要先取log再做分析的。
options(timeout = 99999999)
getwd()
rm(list = ls())
library(GEOquery)
library(stringr)
gse <- "GSE26440"
# 1.获得表达矩阵 ----------------------------------------------------------------
eSet1 <- getGEO(gse,
destdir = '.',
getGPL = F)
#(1)提取表达矩阵exp
exp1 <- exprs(eSet1[[1]])
exp1[1:4,1:4]
range(exp1)
# [1] 0.01 1208.00
exp1 <- log2(exp1+1)
range(exp1)
有点惊讶这样的错误编辑竟然没有发现,而且还大大方方地出现在文章正文中,哪怕作为补充材料出现都值得质疑吧?~
PS:我们并不是为了针对文章作者,而是仅就文章中的问题作出合理的质疑。当然,我们的观点可能并不正确,希望大家从学术讨论的角度出发 peace & love ?