? Monocle 3 | 太牛了!单细胞必学R包!~(二)(寻找marker及注释细胞)
? Monocle 3 | 太牛了!单细胞必学R包!~(二)(寻找marker及注释细胞)

1写在前面
昨天又是不睡觉的一天,晚上还被家属讲了一通,理由是我去急诊了,没有在办公室待着,他老公疼没人去看。?
我的解释是只有我一个值班医生,不可能那么及时,而且也不是什么急症啊。?
好的,说完家属就暴雷了,“这么大的医院就安排一个大夫值班吗!?”?
我也是懒得跟她battle,这个世界不是谁生病就谁有理吧!~?
今天继续接上一次monocle3的教程吧。?
2用到的包
library(tidyverse)
library(monocle3)
library(garnett)
3示例数据
还是上一次运行完的结果哦,不知道大家有没有保存。?
cds
4寻找marker genes
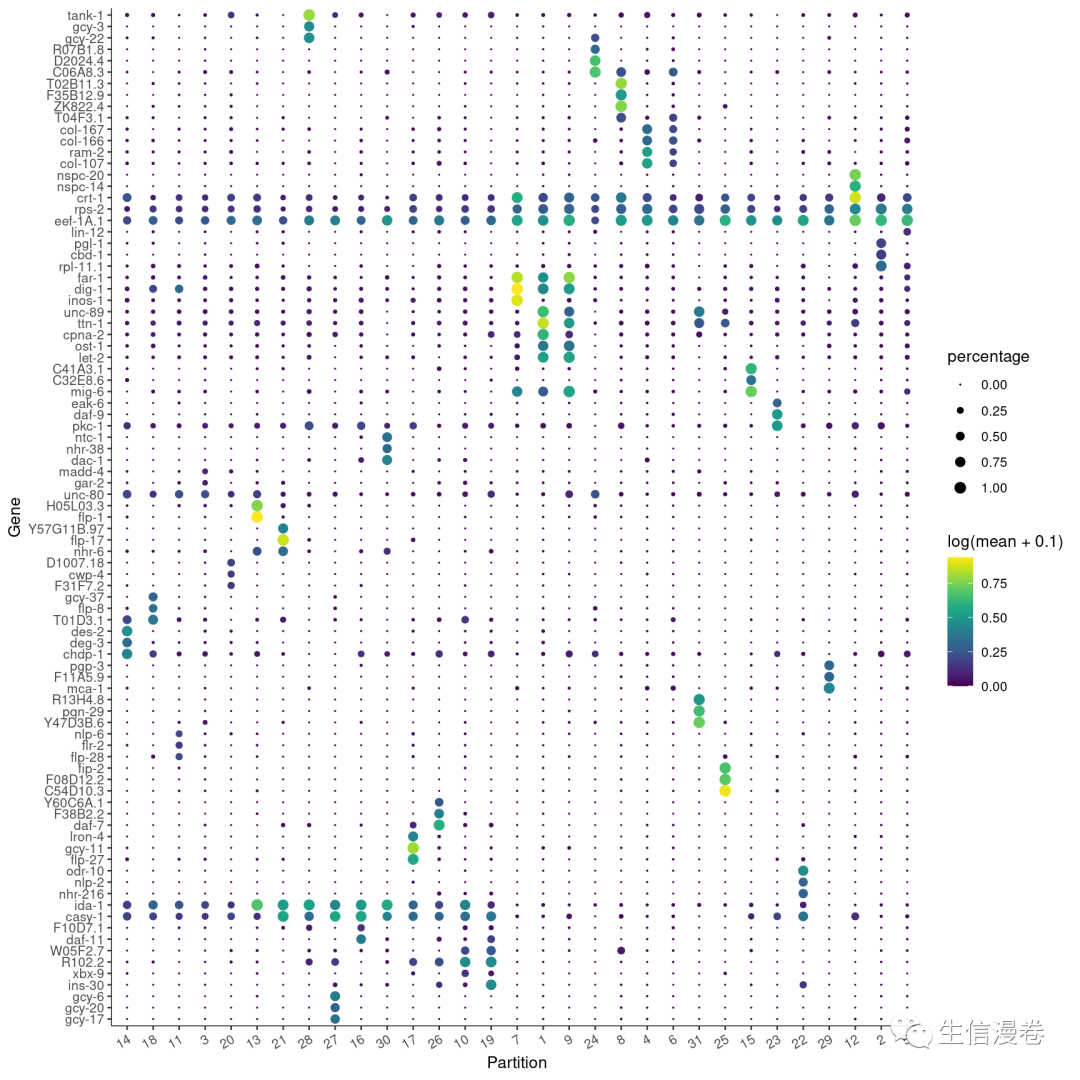
完成细胞聚类后,我们就可以使用top_markers()来选中marker genes。?
marker_test_res <- top_markers(cds, group_cells_by="partition",
reference_cells=1000, cores=8)
接着我们就可以根据cluster,partition或者colData(cds)来确定top n的基因表达量。?
top_specific_markers <- marker_test_res %>%
filter(fraction_expressing >= 0.10) %>%
group_by(cell_group) %>%
top_n(1, pseudo_R2)
top_specific_marker_ids <- unique(top_specific_markers %>% pull(gene_id))
可视化!~?
plot_genes_by_group(cds,
top_specific_marker_ids,
group_cells_by="partition",
ordering_type="maximal_on_diag",
max.size=3)

我们再试试看看top 3的基因。?
top_specific_markers <- marker_test_res %>%
filter(fraction_expressing >= 0.10) %>%
group_by(cell_group) %>%
top_n(3, pseudo_R2)
top_specific_marker_ids <- unique(top_specific_markers %>% pull(gene_id))
plot_genes_by_group(cds,
top_specific_marker_ids,
group_cells_by="partition",
ordering_type="cluster_row_col",
max.size=3)
5注释细胞类型
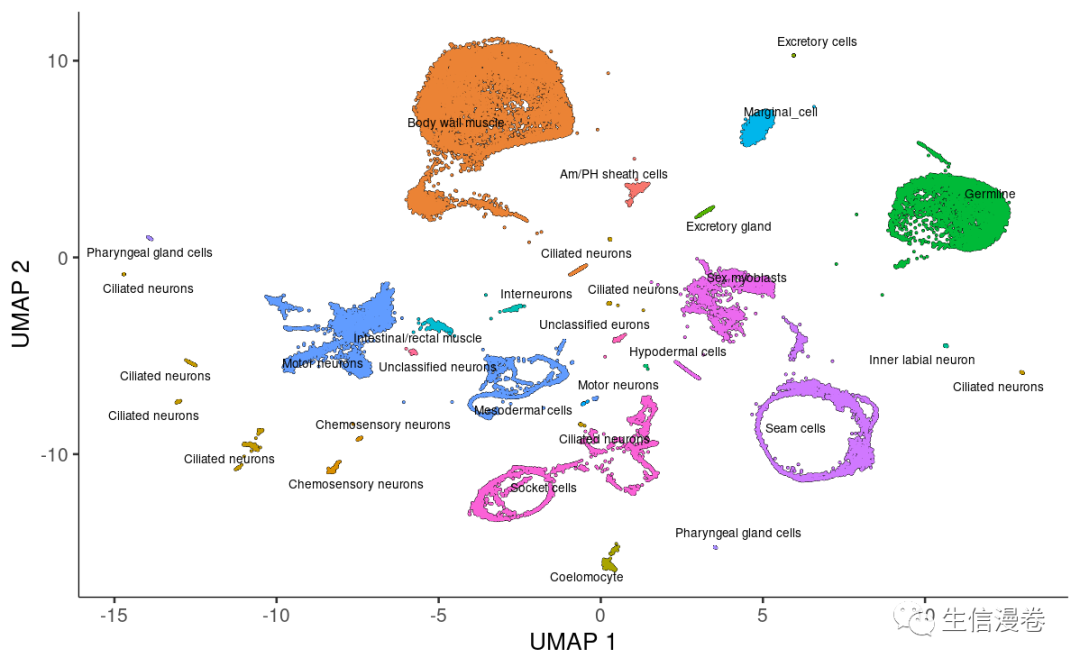
确定每个cluster的类型对许多下游分析至关重要。?
常用的方法是首先对细胞进行聚类,然后根据其基因表达谱为每个cluster注释一个细胞类型。?
我们先创建一列新的colData。?
colData(cds)$assigned_cell_type <- as.character(partitions(cds))
然后我们进行一下手动注释,其实手动注释是最准的。?
colData(cds)$assigned_cell_type <- dplyr::recode(colData(cds)$assigned_cell_type,
"1"="Body wall muscle",
"2"="Germline",
"3"="Motor neurons",
"4"="Seam cells",
"5"="Sex myoblasts",
"6"="Socket cells",
"7"="Marginal_cell",
"8"="Coelomocyte",
"9"="Am/PH sheath cells",
"10"="Ciliated neurons",
"11"="Intestinal/rectal muscle",
"12"="Excretory gland",
"13"="Chemosensory neurons",
"14"="Interneurons",
"15"="Unclassified eurons",
"16"="Ciliated neurons",
"17"="Pharyngeal gland cells",
"18"="Unclassified neurons",
"19"="Chemosensory neurons",
"20"="Ciliated neurons",
"21"="Ciliated neurons",
"22"="Inner labial neuron",
"23"="Ciliated neurons",
"24"="Ciliated neurons",
"25"="Ciliated neurons",
"26"="Hypodermal cells",
"27"="Mesodermal cells",
"28"="Motor neurons",
"29"="Pharyngeal gland cells",
"30"="Ciliated neurons",
"31"="Excretory cells",
"32"="Amphid neuron",
"33"="Pharyngeal muscle")
可视化咯!~?
plot_cells(cds, group_cells_by="partition", color_cells_by="assigned_cell_type")
我们可以利用choose_cells深入解析某个cluster中的亚群。?
手动选择就行了,非常简单。?
cds_subset <- choose_cells(cds)
这个时候我们需要用到graph_test()取了解亚群之间的表达差异。?
pr_graph_test_res <- graph_test(cds_subset,
neighbor_graph= "knn", #"knn", "principal_graph",
cores=8)
pr_deg_ids <- row.names(subset(pr_graph_test_res, morans_I > 0.01 & q_value < 0.05))
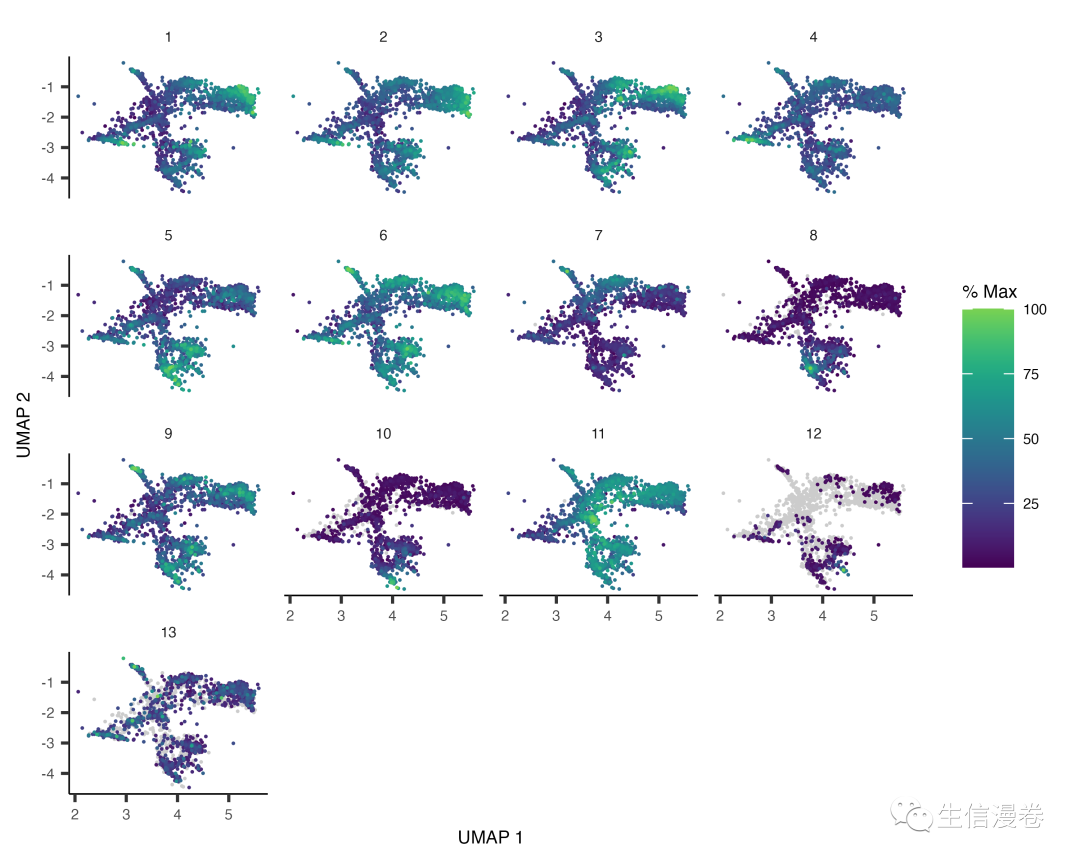
我们甚至可以把其中表达相似的基因归为一个module。?
gene_module_df <- find_gene_modules(cds_subset[pr_deg_ids,], resolution=1e-3)
可视化module表达。?
plot_cells(cds_subset,
genes=gene_module_df,
show_trajectory_graph=F,
label_cell_groups=F)
我们试试把分辨率再提高一些!~?
cds_subset <- cluster_cells(cds_subset, resolution=1e-2)
plot_cells(cds_subset, color_cells_by="cluster")
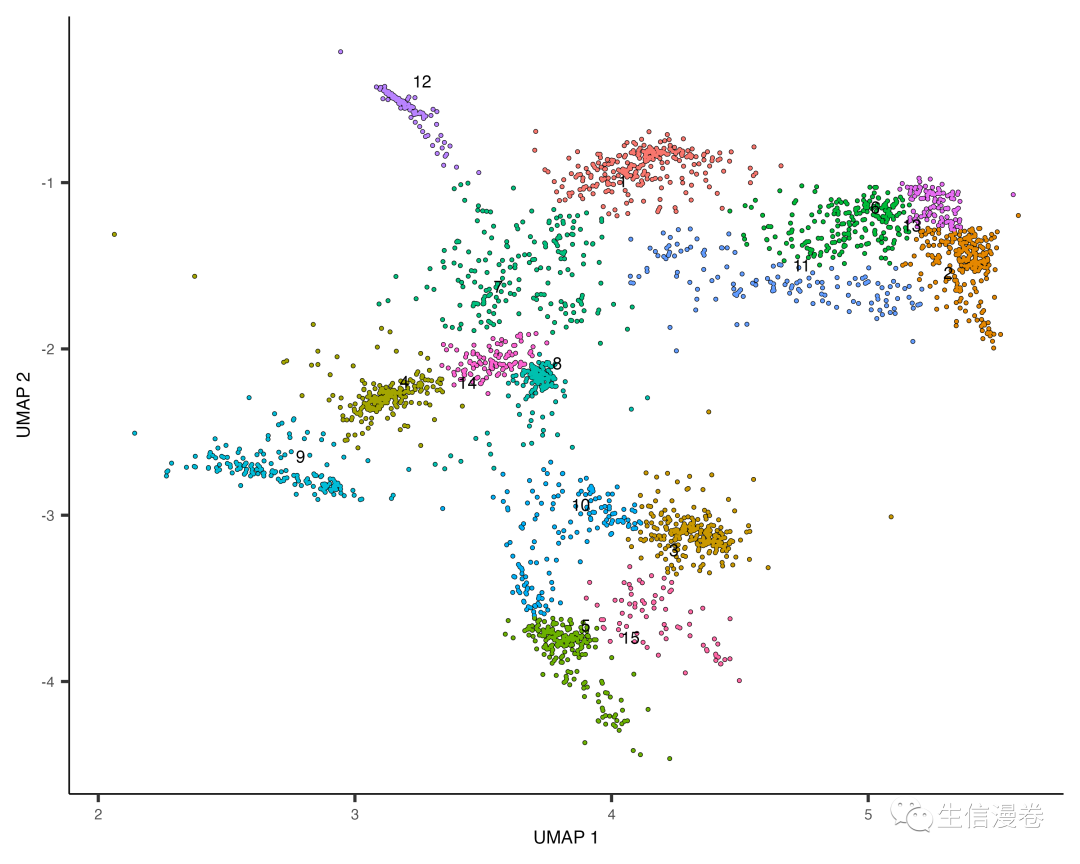
colData(cds_subset)$assigned_cell_type <- as.character(clusters(cds_subset)[colnames(cds_subset)])
colData(cds_subset)$assigned_cell_type <- dplyr::recode(colData(cds_subset)$assigned_cell_type,
"1"="Sex myoblasts",
"2"="Somatic gonad precursors",
"3"="Vulval precursors",
"4"="Sex myoblasts",
"5"="Vulval precursors",
"6"="Somatic gonad precursors",
"7"="Sex myoblasts",
"8"="Sex myoblasts",
"9"="Ciliated neurons",
"10"="Vulval precursors",
"11"="Somatic gonad precursor",
"12"="Distal tip cells",
"13"="Somatic gonad precursor",
"14"="Sex myoblasts",
"15"="Vulval precursors")
plot_cells(cds_subset, group_cells_by="cluster", color_cells_by="assigned_cell_type")
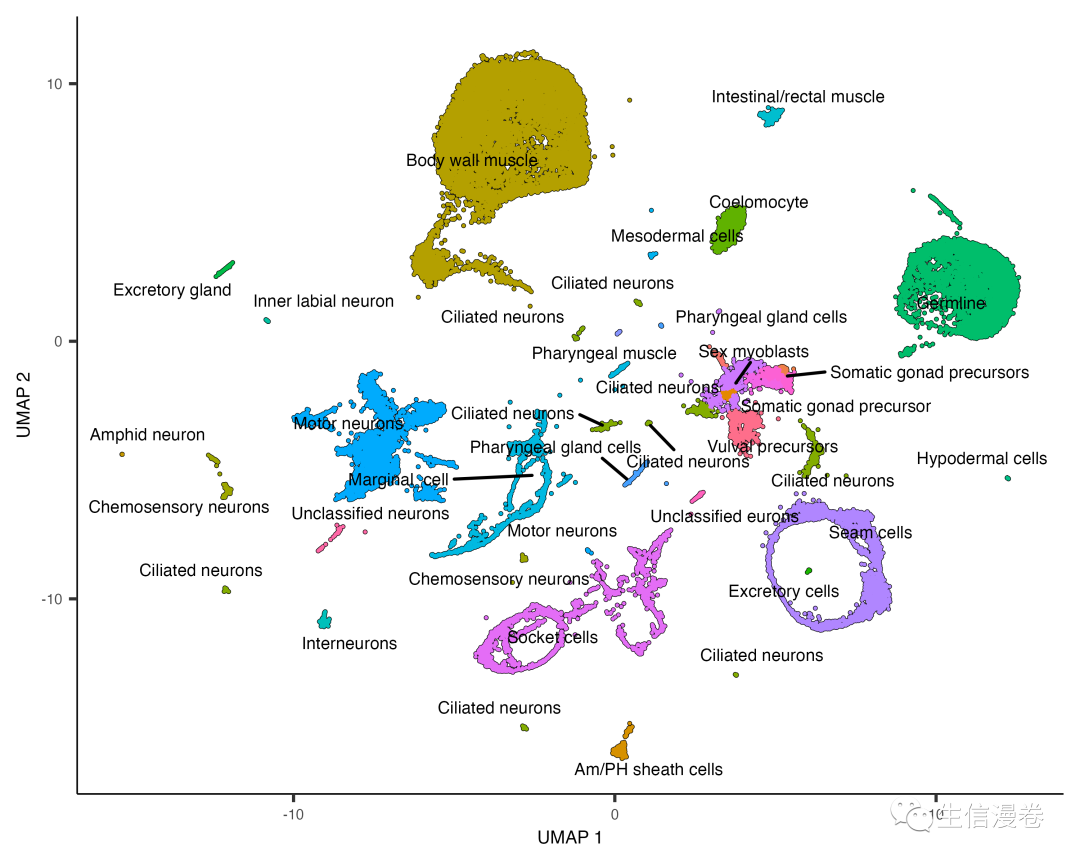
colData(cds)[colnames(cds_subset),]$assigned_cell_type <- colData(cds_subset)$assigned_cell_type
cds <- cds[,colData(cds)$assigned_cell_type != "Failed QC" | is.na(colData(cds)$assigned_cell_type )]
plot_cells(cds, group_cells_by="partition",
color_cells_by="assigned_cell_type",
labels_per_group=5)
6自动注释
这里我们用到的是Garnett。?
首先我们需要确定top_markers。?
assigned_type_marker_test_res <- top_markers(cds,
group_cells_by="assigned_cell_type",
reference_cells=1000,
cores=8)
接着过滤一下marker genes。?
过滤条件:?
1?? JS specificty score > 0.5;
2?? logistic test 有意义;
3?? 不是多个细胞类型的marker。
garnett_markers <- assigned_type_marker_test_res %>%
filter(marker_test_q_value < 0.01 & specificity >= 0.5) %>%
group_by(cell_group) %>%
top_n(5, marker_score)
garnett_markers <- garnett_markers %>%
group_by(gene_short_name) %>%
filter(n() == 1)
然后会生成一个marker文件。?
这个文件你也可以进一步的加工一下,根据你的生物学背景等。?
generate_garnett_marker_file(garnett_markers, file="./marker_file.txt")
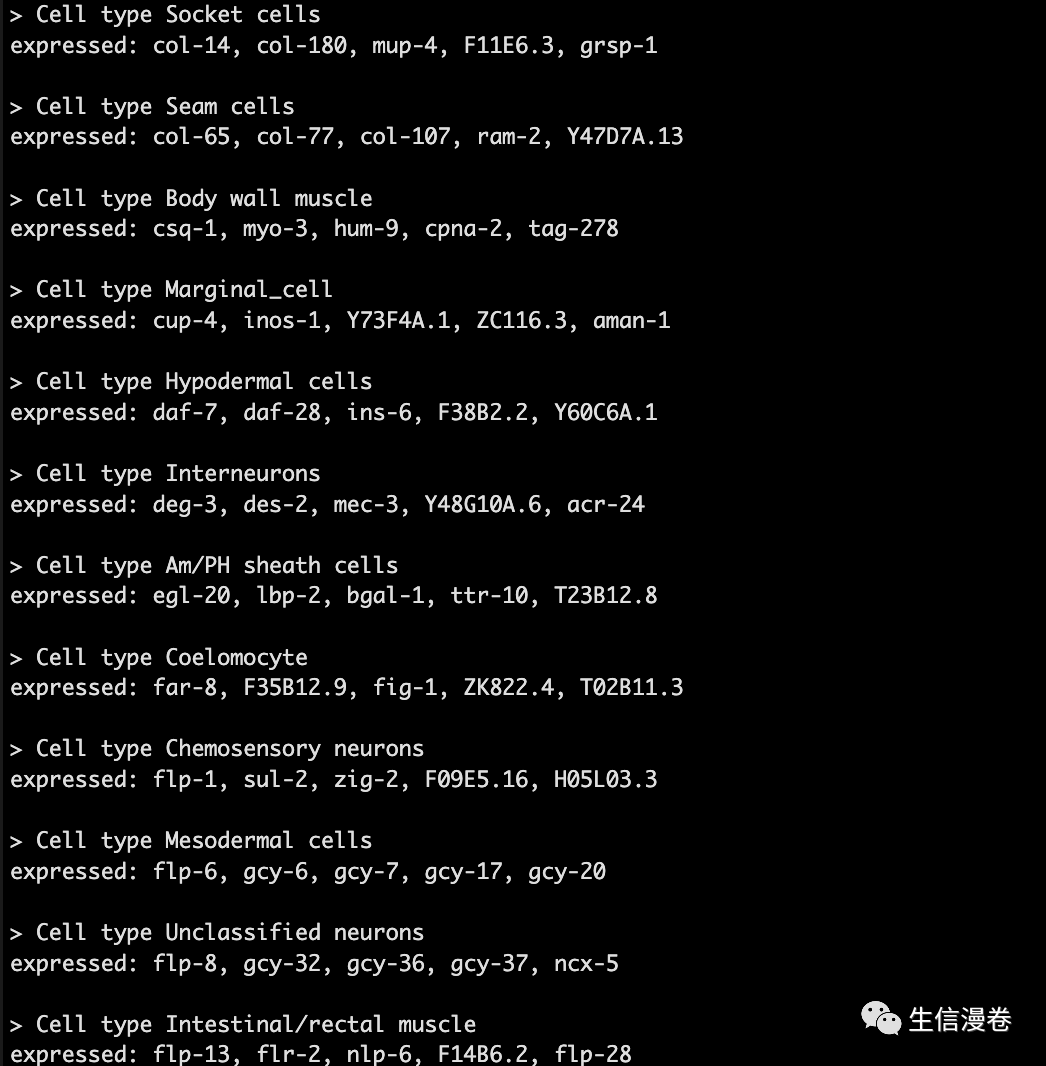
现在可以注释咯,我们先训练一下classifier。?
colData(cds)$garnett_cluster <- clusters(cds)
worm_classifier <- train_cell_classifier(cds = cds,
marker_file = "./marker_file.txt",
db= org.Ce.eg.db::org.Ce.eg.db,
cds_gene_id_type = "ENSEMBL",
num_unknown = 50,
marker_file_gene_id_type = "SYMBOL",
cores=8)

现在可以使用前面训练好的worm_classifier了。?
cds <- classify_cells(cds, worm_classifier,
db = org.Ce.eg.db::org.Ce.eg.db,
cluster_extend = TRUE,
cds_gene_id_type = "ENSEMBL")
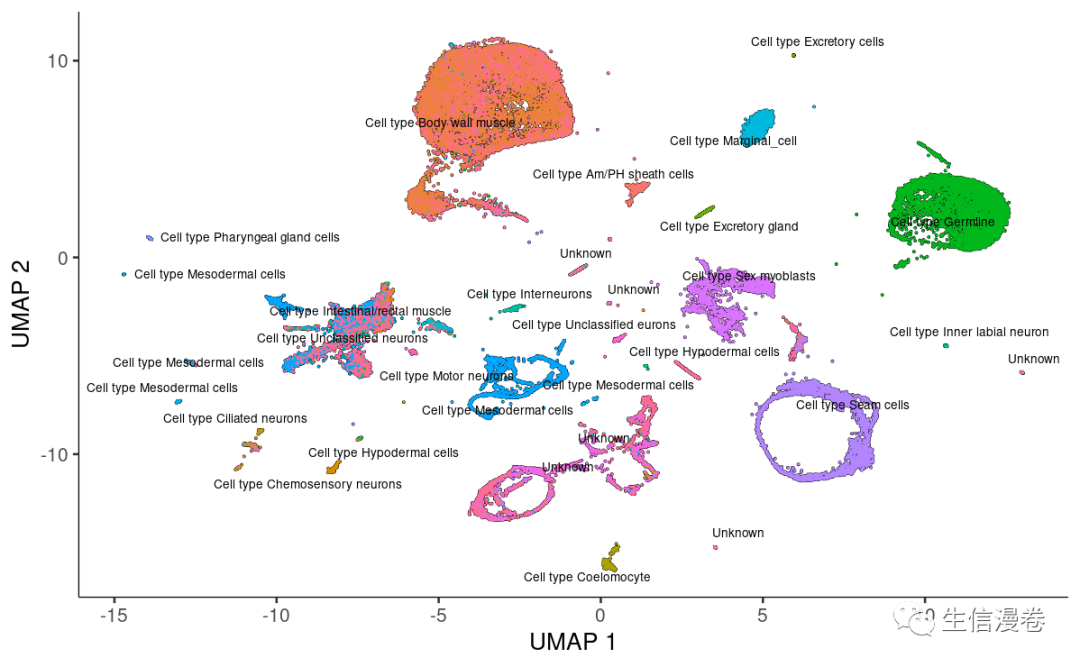
可视化一下,果然还是挺适合我等懒?的!~?
plot_cells(cds,
group_cells_by="partition",
color_cells_by="cluster_ext_type")
线虫的现成模型,不需要自己训练了:?
ceWhole <- readRDS(url("https://cole-trapnell-lab.github.io/garnett/classifiers/ceWhole_20191017.RDS"))
cds <- classify_cells(cds, ceWhole,
db = org.Ce.eg.db,
cluster_extend = TRUE,
cds_gene_id_type = "ENSEMBL")