都是百万单细胞起步了吗

交流群有小伙伴提问不知道如何处理130万这个数量级的单细胞转录组数据,让我感觉很有意思,因为普通人是很难接触到这个数据量的单细胞项目,如果有这个实力那么理所当然知道如何处理不至于来微信交流群问。。。。
130万这个数量级的单细胞转录组数据
也就是说,普通人其实并没有这个能力也不需要面临这样的130万这个数量级的单细胞转录组数据的烦恼!我们拿这个HRA002184数据集举例:
- https://ngdc.cncb.ac.cn/gsa-human/browse/HRA002184
它对应的文章是:《Single-cell analyses implicate ascites in remodeling the ecosystems of primary and metastatic tumors in ovarian cancer》,数据集页面就描述的很清楚, 是:225,373 high-quality cell profiles from primary ovarian tumour, peritoneal metastasis tumour, pelvic lymph node, malignant ascites, and peripheral blood mononuclear cell (PBMC) samples.
页面的 个体和样本信息 显示的这个研究是14个病人的39个样品,如下所示:
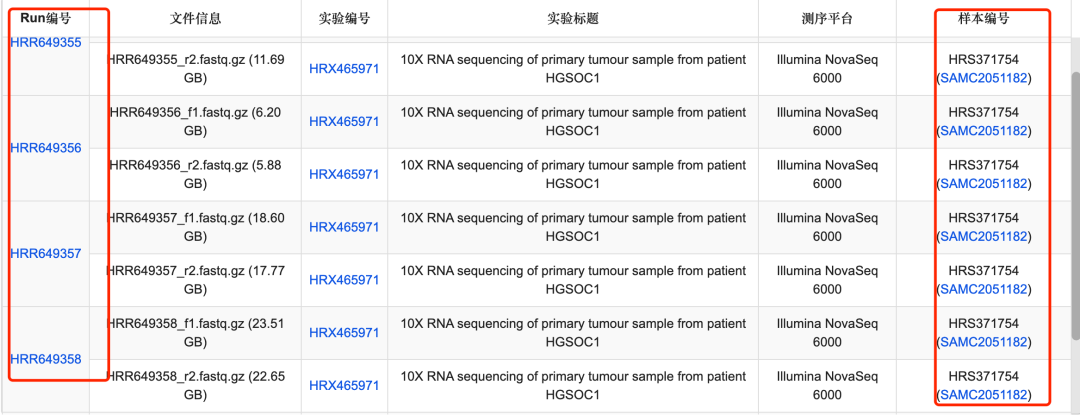
14个病人的39个样品
这样的样品数量,拿到225,373 的细胞数量是合理的,因为每个10x技术的单细胞转录组样品都应该是5到8千的细胞数量。那么为什么有人会以为这个研究有130万这个数量级的单细胞转录组数据,因为初学者可能会不太懂单细胞的测序数据结构。以为页面里面的run就是每个10x样品,其实是每个样品可以有多个run
关于国家基因组科学数据中心
国家基因组科学数据中心(National Genomics Data Center, NGDC)的人类遗传资源组学原始数据归档库(Genome Sequence Archive for Human, GSA-Human)是一个用于收集、存储、管理和共享人类遗传资源原始数据的数据库。在GSA-Human中,数据格式和ID标识遵循一定的规范,以确保数据的准确性和可追溯性。
- HRI ID:HRI代表“Human Research Individual”,即个体的标识符。每个在GSA-Human数据库中存储的个体或样本都会有一个唯一的HRI ID,用于标识特定的个体或样本。
- HRS ID:HRS代表“Human Research Sample”,即样本的标识符。与HRI ID类似,HRS ID用于唯一标识一个样本。
- HRR ID:HRR代表“Human Research Run”,可能指的是测序运行的标识符。它可能用于标识特定的测序实验或测序批次。
- HRX ID:HRX的具体含义在提供的文档中没有明确说明,但根据上下文推测,它可能代表与人类遗传资源相关的某种特定数据集或实验的标识符。
如下所示可以看到每个HRS的编号都会对应很多HRR的编号 :
所以,虽然说我们可以看到 926个fq文件,其实对应的是463个run,因为每个run在这个作者上传的fq1和fq2文件,如下所示:
ls -lh */*gz|cut -d" " -f 6-|head
2.9G Mar 26 17:26 HRR649563/HRR649563_S1_L001_R1_001.fastq.gz
11G Mar 26 17:25 HRR649563/HRR649563_S1_L001_R2_001.fastq.gz
3.1G Mar 26 21:32 HRR649564/HRR649564_S1_L001_R1_001.fastq.gz
12G Mar 26 21:31 HRR649564/HRR649564_S1_L001_R2_001.fastq.gz
2.5G Mar 26 15:16 HRR649565/HRR649565_S1_L001_R1_001.fastq.gz
9.4G Mar 26 15:18 HRR649565/HRR649565_S1_L001_R2_001.fastq.gz
2.5G Mar 26 10:58 HRR649566/HRR649566_S1_L001_R1_001.fastq.gz
9.6G Mar 26 10:58 HRR649566/HRR649566_S1_L001_R2_001.fastq.gz
139M Mar 26 12:41 HRR649567/HRR649567_S1_L001_R1_001.fastq.gz
499M Mar 26 12:41 HRR649567/HRR649567_S1_L001_R2_001.fastq.gz
但是这个463个run,对应的很明显就39个样品,所以实际上在修改他们的名字的时候,就不能是采用上面的规则。。。。
而且从文件大小也可以看出来的,上面的每个run的fq1和fq2文件都不大啊,如果是真正的10x单细胞转录组,比如:PRJCA007562,可以看到每个样品的fq1和fq2文件是很大的:
12G 8月 31 12:48 HRR568828_f1.fastq.gz
44G 8月 31 18:38 HRR568828_r2.fastq.gz
18G 8月 31 13:32 HRR568829_f1.fastq.gz
70G 8月 31 23:18 HRR568829_r2.fastq.gz
47G 9月 1 01:05 HRR568830_r2.fastq.gz
13G 8月 31 14:17 HRR568831_f1.fastq.gz
47G 9月 1 01:35 HRR568831_r2.fastq.gz
这样的10x单细胞转录组技术,必须是以样品为单位的修改名字哦!比如我们前面介绍的不知道10x单细胞转录组样品和fastq文件的对应关系,就是多个run的fq文件修改为样品开头的文件:
7_S1_L001_R1_001.fastq.gz -> /home/PRJNA762594/SRR15860132_1.fastq.gz
7_S1_L001_R2_001.fastq.gz -> /home/PRJNA762594/SRR15860132_2.fastq.gz
7_S1_L002_R1_001.fastq.gz -> /home/PRJNA762594/SRR15860143_1.fastq.gz
7_S1_L002_R2_001.fastq.gz -> /home/PRJNA762594/SRR15860143_2.fastq.gz
7_S1_L003_R1_001.fastq.gz -> /home/PRJNA762594/SRR15860154_1.fastq.gz
7_S1_L003_R2_001.fastq.gz -> /home/PRJNA762594/SRR15860154_2.fastq.gz
7_S1_L004_R1_001.fastq.gz -> /home/PRJNA762594/SRR15860155_1.fastq.gz
7_S1_L004_R2_001.fastq.gz -> /home/PRJNA762594/SRR15860155_2.fastq.gz
一个样品可以有三五个run,甚至可以有成百上千个run,但是它仍然是一个样品只能说是跑一次cellranger得到一个表达量矩阵哦。在处理fastq文件时,如果遇到多个样品或多个测序通道的情况,需要仔细地按照fastq文件的命名规则进行整理和重命名,以确保数据分析的准确性。比如我在10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元) 演示的:
SIGAA11_S40_L007_R1_001.fastq.gz
SIGAA11_S40_L007_R2_001.fastq.gz
SIGAA11_S40_L007_I1_001.fastq.gz
SIGAA11_S40_L008_R1_001.fastq.gz
SIGAA11_S40_L008_R2_001.fastq.gz
SIGAA11_S40_L008_I1_001.fastq.gz
SIGAA11_S40_L009_R1_001.fastq.gz
SIGAA11_S40_L009_R2_001.fastq.gz
SIGAA11_S40_L009_I1_001.fastq.gz
针对10x单细胞转录组的fastq文件,其命名通常遵循一定的规则,这些规则有助于识别样品的来源和测序过程中的不同阶段。根据您提供的文件名示例,我们可以分析出以下规律:
- 样品标识:
SIGAA11通常表示特定的样品名称或ID,用于区分不同的样品。 - 样品序号:
S40表示该样品在测序运行中的序号。在10X技术中,S后面的数字通常用来标识样品的唯一性,特别是在一个实验中处理多个样品时。 - 库标识:
L009表示该样品被分配到的库(Library)编号。在一次测序运行中,一个样品可能会被分成多个库进行测序,以增加测序深度或处理样品量。 - 测序通道:
R1和R2表示测序的两个独立通道。在10X单细胞转录组测序中,R1通常代表细胞barcode和UMI(Unique Molecular Identifier),而R2代表实际的cDNA序列。 - 索引文件:
I1有时候也会出现在文件名中,它通常包含了测序的索引信息,但在某些情况下,如第一个搜索结果3所述,I1文件理论上可以抛弃,因为所需的信息已经在R1和R2中包含了。 - 文件格式:
.fastq.gz表示这是一个经过压缩的fastq格式文件。Fastq格式是一种文本文件,包含了测序得到的核酸序列及其质量分数。 - 文件后缀:
_001这通常表示该文件是该样品测序数据的一部分,有时一个样品的测序数据会分成多个文件存储,后缀用来区分这些文件。
在处理这些fastq文件时,需要确保文件名的一致性和正确性,以便在后续的数据分析流程中,如使用Cell Ranger软件进行单细胞转录组数据分析时,能够正确地识别和处理这些数据。
只需要是合理的修改好名字,正常走cellranger的定量流程即可,代码我已经是多次分享了。参考:
- 10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元)
- 10X的单细胞转录组原始数据也可以在EBI下载
- 一个10x单细胞转录组项目从fastq到细胞亚群
- 一文打通单细胞上游:从软件部署到上游分析
- PRJNA713302这个10x单细胞fastq实战
- 一次曲折且昂贵的单细胞公共数据获取与上游处理
- 只能下载bam文件的10x单细胞转录组项目数据处理
- 不知道10x单细胞转录组样品和fastq文件的对应关系
- 10X单细胞转录组测序数据的 SRA转fastq踩坑那些事
- 10x的单细胞转录组fastq文件的R1和R2不能弄混哦
最后可以得到如下所示的每个样品的表达量矩阵文件以及对应的报表:
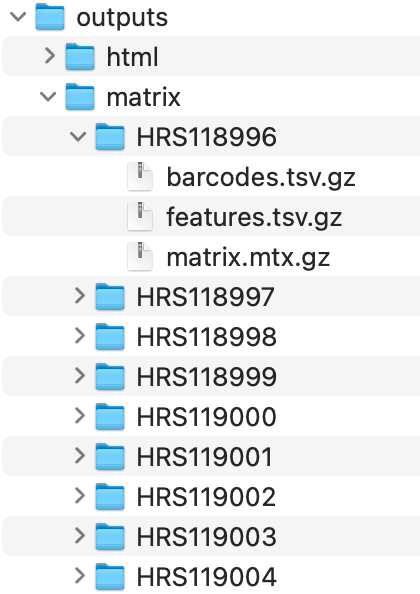
每个样品的表达量矩阵文件以及对应的报表
也就是说,上面的研究是14个病人的39个样品,所以理论上就应该是出来39个文件夹而已。也就是22.5万个细胞而已。